Wiedemann‑Steiner syndrome in a 2‑year‑old patient due to a rare nonsense KMT2A mutation of de novo origin: A case report
- Authors:
- Published online on: February 5, 2024 https://doi.org/10.3892/ije.2024.20
- Article Number: 1
-
Copyright : © Keramida et al. This is an open access article distributed under the terms of Creative Commons Attribution License [CC BY 4.0].
Abstract
Introduction
Wiedemann-Steiner syndrome (WSS) is a genetic disorder characterized by a wide range of clinical symptoms, including developmental delay, intellectual disability, distinctive facial features, and other clinical features (1). Some of the facial features are thick eyebrows with lateral flare, vertically narrow and downward slanted palpebral fissures, widely spaced eyes (hypertelorism), long eyelashes, wide nasal bridge, broad nasal tip, thin vermilion of the upper lip and thick scalp hair (1,2). In addition to the aforementioned clinical manifestations, other features include ophthalmologic anomalies, hand anomalies (like brachydactyly and clinodactyly), congenital heart defects, as well as prenatal and postnatal growth restriction (1,2).
WSS is part of a group of disorders known as chromatinopathies, which are caused by mutations in genes that encode components of the epigenetic machinery (3). More specifically, WSS has been linked to heterozygous pathogenic mutations in the lysine methyltransferase 2A (KMT2A) gene (1,4). KMT2A encodes the protein lysine methyltransferase 2A, which is part of the KMT family. This group of proteins is part of the epigenetic machinery and is crucial for gene expression. More specifically, the KMT family catalyzes the transfer of methyl groups from S-adenosylmethionine to lysine residues on histone H3 tails. KMT2A in particular, is responsible for transcriptional activation through lysine 4 of histone 3 (H3K4) methylation. H3K4 methylation positively regulates the transcription of multiple genes, including genes involved in hematopoiesis and neuronal development (5).
The present study reports the case of a 2-year-old female patient that presented with a variety of clinical features including hypertelorism, thick eyebrows and epicanthus. Whole exome sequencing (WES) analysis was performed and the results revealed the presence of a heterozygous pathogenic mutation in KMT2A, namely c.517C>T, suggesting a WSS diagnosis. Its de novo origin was confirmed by DNA analysis of the parents. This is a rare mutation that has been documented only twice in the ClinVar database and to the best of our knowledge, there are no reported cases of this mutation in the scientific literature. Hence, to the best of our knowledge, the present study describes the first report of WSS caused by the c.517C>T mutation in KMT2A, that includes a detailed description of the clinical manifestations in the patient.
Case report
A 2-year-old girl was referred to Access to Genome, Clinical Laboratory Genetics for genetic testing due to a variety of clinical features. More specifically, the girl exhibited Kabuki-like gestalt with long palpebral fissures, high frontal hairline, bilateral severe epicanthus, hypertelorism, accentuated eyebrows with medial flare, a thin upper lip, bulbous nose, posteriorly rotated ears with anteverted lobuli, hypoplastic nipples, increased intermamillary distance, hypotonic kyphosis, mild umbilical hernia, joint laxity, hypotonia, mild dorsal hypertrichosis, early tooth eruption, thin lower legs with protruding knees and protruding heels without rocker bottom. She did not exhibit a delay in gross and fine motor development and language comprehension; however, she had delayed expressive language. Moreover, failure to thrive was observed, despite good feeding behavior. The patient was hyperkinetic, but without attention deficit. Moreover, an electroencephalogram revealed normal results, renal, abdominal and heart ultrasound results were normal, antigliadin antibody tests were negative, hearing test results were normal, sweat test results were normal, and hematological, biochemical and endocrinological test results were also normal. An ophthalmological investigation at 18 months of age revealed some immaturity of the optic nerve. At 24 months of age, her weight was 10 kg, between 3rd and 25th percentile. She had a normal female karyotype (46,XX).
WES analysis was performed on the DNA of the patient that was isolated from whole blood cells. Exome amplification was performed using AmpliSeq Exome RDY (Thermo Fisher Scientific, Inc.). Nucleotide sequencing was performed using the Ion Chef Instrument in combination with the Ion GeneStudio S5 System (Thermo Fisher Scientific, Inc.). Subsequently, 4,432 genes associated with known genetic diseases and syndromes were analyzed. Data evaluation and interpretation were based on the clinical features of the patient. The analysis was performed using Alamut Visual and Varsome Clinical (Saphetor SA) bioinformatic analysis systems. All findings from the aforementioned analysis were evaluated in accordance with the international literature and the American College of Medical Genetics and Genomics (ACMG) guidelines (6). The reference genome was UCSC hg19. Bioinformatics analysis revealed the presence of a heterozygous mutation in KMT2A, namely c.517C>T. This is a nonsense mutation (p.Arg173Ter or R173*) that leads to a premature stop codon in exon 3.
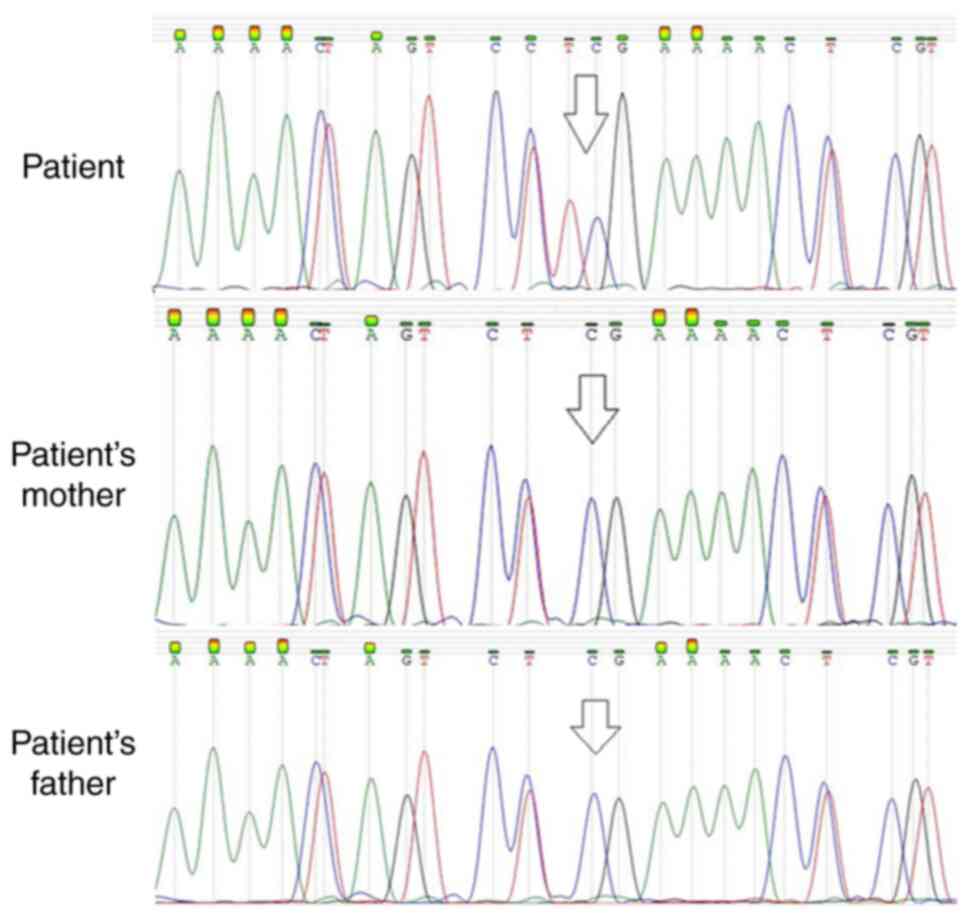
Sanger sequencing was performed in the patient in order to confirm the presence of the c.517C>T mutation. Following DNA amplification using PCR, part of the KMT2A gene was sequenced and was compared to the control sequence. The sequences of the primers used were: ACTCAAGTTGAACTCAGTACAAAATGG (forward primer) and CTTTCTTCTTGATCTTATCTCCAGATTTGGT (reverse primer). The results confirmed the presence of the rare mutation. In order to establish whether the mutation was inherited or whether it occurred de novo, Sanger sequencing of this part of KMT2A was performed on parental DNA that was isolated from whole blood cells. The mutation was not present in either of the patient's parents, indicating that this was a de novo mutation (Fig. 1).
Discussion
The present study describes the case of a 2-year-old female patient with a phenotype characterized by hypertelorism, thick eyebrows, epicanthus, dysplastic ears and others. WES analysis revealed that she is a carrier of the c.517C>T nonsense mutation in KMT2A and further analysis of her parents revealed that this mutation occurred de novo. The clinical features of the patient combined with the identification of the KTM2A mutation are supportive of a WSS diagnosis.
The KMT2A protein is a component of the epigenetic machinery, playing a crucial role in epigenetic transcriptional activation (4). KMT2A is an evolutionary conserved gene that is critical for various functional processes during embryonic development, spanning from hematopoiesis to neurogenesis (4). The encoded protein mediates chromatin modifications associated with epigenetic transcriptional activation and functions as a positive regulator for the expression of numerous target genes (2). Included in those genes are genes that belong to the Hox complex, as well as other genes involved in embryonic development (2,7). The disruption of KMT2A leads to the dysfunction of the epigenetic machinery and the transcriptional activation of genes that are critical for development. This ultimately leads to the manifestation of clinical symptoms associated with WSS (7).
The role of KMTA has been demonstrated in murine and zebrafish animal models. For instance, a previous study on zebrafish has demonstrated that KMT2A is essential for neural development in zebrafish embryos (8). Moreover, previous research has demonstrated that the complete disruption of KMT2A in mouse embryos is lethal, while heterozygous animals exhibit a variety of symptoms, including growth delay and skeletal malformations (9). The phenotypical differences between the homozygous and the heterozygous animals suggest a dosage-sensitive regulation by the KMT2A protein (8). Moreover, the essential role of KMT2A in neurogenesis was demonstrated in a study where impaired neuronal differentiation in the postnatal mouse brain was observed in KMT2A knockout mice (5). Other studies on mice have also demonstrated that there is a high expression of KMT2A in adult hippocampal neurons and that KMT2A is vital for synaptic plasticity, cognition, complex behaviors and long-term memory (10,11).
Mutations in KMT2A have been observed throughout the gene, which consists of 37 exons. However, a pathogenic mutation hotspot in exon 27 exists. The majority of the observed mutations lead to the loss of function of KMT2A (4). The c.517C>T mutation is a nonsense mutation, which leads to a premature stop codon in exon 3. Nonsense mutations in KMT2A are a known pathogenicity mechanism for the KMT2A gene. More specifically, these mutations lead to the nonsense mediated decay of the transcript, which causes haploinsufficiency, ultimately leading to the clinical features associated with WSS (1,12). Notably, the c.517C>T mutation has not yet been reported in the gnomAD database and only two submissions exist in the ClinVar database. Moreover, to date, to the best of our knowledge, there is no report of this variant in the scientific literature. According to the ACMG guidelines, this variant is categorized as ‘Pathogenic’ based on the PVS1, PM2, PP5 and PM6 criteria (6).
The clinical features of WSS overlap with those of certain other syndromes (1). Due to the wide range of phenotypic characteristics of WSS, differential diagnosis based on the observed phenotype alone is difficult (2). Notably, mutations in KMT2A have been identified in cases initially diagnosed as Coffin-Siris syndrome, Cornelia de Lange syndrome, Kabuki syndrome and Rubinstein-Taybi syndrome (13-17), which are also chromatinopathies and share similar clinical manifestations. In instances where patients exhibit characteristics suggestive of a chromatinopathy, WES serves as an aid in the differential diagnosis within this group of disorders.
In the case described herein, a de novo KMT2A mutation was identified in the patient. This mutation is a nonsense mutation, resulting in a premature stop codon, a known pathogenicity mechanism for this gene. Additionally, the symptoms observed in the patient align with the expected consequences of this mutation. Based on the aforementioned information, it can be inferred that the detected KMT2A mutation is the underlying cause of the patient's condition. To the best of our knowledge, this is the first report of WSS syndrome caused by the c.517C>T mutation in KMT2A. The present study provides a detailed description of the phenotypic characteristics of the patient that is critical for genetic counseling in cases of prenatal or postnatal detection of this mutation. Moreover, the case description in the present study may be valuable for evaluating other patients who exhibit features of a chromatinopathy. Finally, the present study emphasizes the importance of utilizing WES for achieving a differential diagnosis when a chromatinopathy is suspected.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
CK substantially contributed to the design of the study and prepared the manuscript. EM was in charge of patient management and project supervision. EM and IP critically revised the manuscript. IP, ES and CE performed WES and Sanger sequencing. YG was responsible for the evaluation of the patient and genetic counseling. EP, EA and AG were responsible for the medical treatment and assessment of the patient. EM and IP confirm the authenticity of all the raw data. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Written informed consent was obtained from the parents and the patient for the inclusion of their data in the present case report. Any information revealing the patient's identity was not included. All procedures followed were conducted according to The Declaration of Helsinki 1975, as revised in 2008.
Patient consent for publication
Written informed consent was obtained from the patient's parents for publication of the present case report and any accompanying images.
Competing interests
The authors declare that they have no competing interests.
References
|
Sheppard SE and Quintero-Rivera F: Wiedemann-Steiner Syndrome. University of Washington, Seattle, WA, 2022. | |
|
Yu H, Zhang G, Yu S and Wu W: Wiedemann-Steiner Syndrome: Case report and review of literature. Children (Basel). 9(1545)2022.PubMed/NCBI View Article : Google Scholar | |
|
Fahrner JA and Bjornsson HT: Mendelian disorders of the epigenetic machinery: Postnatal malleability and therapeutic prospects. Hum Mol Genet. 28(R2):R254–R264. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Castiglioni S, Di Fede E, Bernardelli C, Lettieri A, Parodi C, Grazioli P, Colombo EA, Ancona S, Milani D, Ottaviano E, et al: KMT2A: Umbrella gene for multiple diseases. Genes (Basel). 13(514)2022.PubMed/NCBI View Article : Google Scholar | |
|
Lim DA, Huang YC, Swigut T, Mirick AL, Garcia-Verdugo JM, Wysocka J, Ernst P and Alvarez-Buylla A: Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature. 458:529–533. 2009.PubMed/NCBI View Article : Google Scholar | |
|
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al: Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Fontana P, Passaretti FF, Maioli M, Cantalupo G, Scarano F and Lonardo F: Clinical and molecular spectrum of Wiedemann-Steiner syndrome, an emerging member of the chromatinopathy family. World J Med Genet. 9:1–11. 2020. | |
|
Huang YC, Shih HY, Lin SJ, Chiu CC, Ma TL, Yeh TH and Cheng YC: The epigenetic factor Kmt2a/Mll1 regulates neural progenitor proliferation and neuronal and glial differentiation. Dev Neurobiol. 75:452–462. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Yu BD, Hess JL, Horning SE, Brown GA and Korsmeyer SJ: Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 378:505–508. 1995.PubMed/NCBI View Article : Google Scholar | |
|
Kim SY, Levenson JM, Korsmeyer S, Sweatt JD and Schumacher A: Developmental regulation of Eed complex composition governs a switch in global histone modification in brain. J Biol Chem. 282:9962–9972. 2007.PubMed/NCBI View Article : Google Scholar | |
|
Jakovcevski M, Ruan H, Shen EY, Dincer A, Javidfar B, Ma Q, Peter CJ, Cheung I, Mitchell AC, Jiang Y, et al: Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and working memory. J Neurosci. 35:5097–5108. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Jones WD, Dafou D, McEntagart M, Woollard WJ, Elmslie FV, Holder-Espinasse M, Irving M, Saggar AK, Smithson S, Trembath RC, et al: De novo mutations in MLL cause wiedemann-steiner syndrome. Am J Hum Genet. 91:358–364. 2012.PubMed/NCBI View Article : Google Scholar | |
|
Bramswig NC, Lüdecke HJ, Alanay Y, Albrecht B, Barthelmie A, Boduroglu K, Braunholz D, Caliebe A, Chrzanowska KH, Czeschik JC, et al: Exome sequencing unravels unexpected differential diagnoses in individuals with the tentative diagnosis of Coffin-Siris and Nicolaides-Baraitser syndromes. Hum Genet. 134:553–568. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Yuan B, Pehlivan D, Karaca E, Patel N, Charng WL, Gambin T, Gonzaga-Jauregui C, Sutton VR, Yesil G, Bozdogan ST, et al: Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest. 125:636–651. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Parenti I, Teresa-Rodrigo ME, Pozojevic J, Ruiz Gil S, Bader I, Braunholz D, Bramswig NC, Gervasini C, Larizza L, Pfeiffer L, et al: Mutations in chromatin regulators functionally link Cornelia de Lange syndrome and clinically overlapping phenotypes. Hum Genet. 136:307–320. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Sobreira N, Brucato M, Zhang L, Ladd-Acosta C, Ongaco C, Romm J, Doheny KF, Mingroni-Netto RC, Bertola D, Kim CA, et al: Patients with a Kabuki syndrome phenotype demonstrate DNA methylation abnormalities. Eur J Hum Genet. 25:1335–1344. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Negri G, Magini P, Milani D, Crippa M, Biamino E, Piccione M, Sotgiu S, Perrìa C, Vitiello G, Frontali M, et al: Exploring by whole exome sequencing patients with initial diagnosis of Rubinstein-Taybi syndrome: The interconnections of epigenetic machinery disorders. Hum Genet. 138:257–269. 2019.PubMed/NCBI View Article : Google Scholar |