
Adipocyte‑rich microenvironment promotes chemoresistance via upregulation of peroxisome proliferator‑activated receptor gamma/ABCG2 in epithelial ovarian cancer
- Authors:
- Published online on: February 27, 2024 https://doi.org/10.3892/ijmm.2024.5361
- Article Number: 37
-
Copyright: © Chen et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Currently, the therapeutic strategy of cancer has shifted from monotherapy to combination therapies, among which chemotherapy still results in marked curative treatment for numerous types of cancer (1). However, chemoresistance is a major obstacle that impedes successful chemotherapeutic treatment. Tumor microenvironment (TME) alterations direct cell states during cellular development, which further drive drug responses (2). The effects of an adipocyte-rich microenvironment (ARM) on chemoresistance have garnered increasing interest (3-6). Thus, there may be value in clarifying the underlying mechanisms and identifying novel targets from the perspective of ARM to improve the efficacy of chemotherapy.
Ovarian cancer (OVCA), a representative adipocyte-rich associated cancer (7,8), is the most lethal gynecological malignancy, with a 5-year survival rate of 48% (9). Of OVCA cases, ~90% are epithelial ovarian cancer (EOC) in terms of histological type and the most common subtype of EOC is high-grade serous ovarian carcinoma (HGSOC; 70% of cases) and it also has the worst prognosis (10). The current chemotherapeutic regimen for EOC is a combination of a platinum compounds and taxane (11). Although most patients initially respond to chemotherapy, 70-80% of the patients relapse and develop multidrug resistance (MDR) against chemotherapy within 2-5 years (12,13). MDR is attributed to a variety of factors, including reduced uptake of drugs by cells, increased efflux and intracellular inactivation (14). In addition, changes in the TME affect cell status and further drive drug responses. Currently, the influence of adipocytes, the primary component of ARM, on chemoresistance in EOC has raised concerns (1,15,16). Adipocytes also secrete certain adipokines, including leptin, visfatin, resistin and adiponectin, amongst other factors, to induce drug resistance (1,15,16). However, little is known regarding the mechanisms involved in the contribution of ARM to chemoresistance.
The human ATP-binding cassette (ABC) transporters, which are responsible for MDR when upregulated (17), serve as the most commonly used pump for drug efflux in an ATP-dependent manner (18,19). ABC transporter G family member 2 (ABCG2), also known as breast cancer resistance protein, plays an important role in MDR by extruding drugs (20,21). Peroxisome proliferator-activated receptor γ (PPARγ), which belongs to the nuclear receptor superfamily, plays a crucial role in the development of adipose tissue and lipid metabolism (11,22). There have been controversial opinions regarding the roles of PPARγ and whether it exhibits an anti-cancer or oncogenic effect (23,24). Currently, little is known regarding the implications of PPARγ in chemoresistance. ABCG2 has been reported to be related to PPARγ activation in dendritic cells (25) and placental choriocarcinoma cells (26). Inhibition of PPARγ suppresses the efflux activity of ABCG2 in M2 macrophages (27). Therefore, the contributions of ARM-educated PPARγ/ABCG2 pathway may highlight novel targets to improve the effects of chemotherapy.
In the present study, it was shown that ARM influenced chemoresistance via a PPARγ/ABCG2 pathway in EOC and whether this mechanism may provide clues for relieving chemoresistance for EOC treatments was assessed.
Materials and methods
Cell lines and reagents
The human HGSOC cell lines OVCAR3 and OVCAR8 were gifts from Professor Zhi Yao (Tianjin Medical University, Tianjin, China) and a mouse ovarian epithelial papillary serous adenocarcinoma cell line ID8 cell was a gift from Professor Luyuan Li (Nankai University, Tianjin, China). All the cells were cultured in RPMI 1640 or DMEM (Thermo Fisher Scientific, Inc.) supplemented with 10% (v/v) FBS (Gibco; Thermo Fisher Scientific, Inc.). The reagents for in vitro studies included cisplatin (DDP; Qilu Pharmaceutical Co., Ltd.) (OVCAR3; 40 μM; ID8; 10 μM); paclitaxel (PTX; Yangtze River Pharmaceutical Group) (OVCAR3; 3 nM; ID8; 20 nM); troglitazone (a PPARγ agonist; Trog, Selleck Chemicals), 25 μM; GW9662 (a PPARγ agonist; Selleck Chemicals), 25 μM; and an ABCG2 inhibitor, KO143 (MedChemExpress), 10 μM. For in vitro studies, cells were pretreated with Trog, GW9662 and KO143 for 2 h after which the media was changed to the normal media for further treatments.
HATES preparation
Sterile peritumoral adipose tissue was dissected from female EOC patients undergoing surgery. HATES were prepared from fresh peritumoral adipose tissue as described previously (28). HATES liquid was collected and frozen at a concentration of 0.8 g/ml with a final concentration of HATES of 0.4 g/ml (diluted with regular medium containing 20% FBS at a ratio of 1:1) for in vitro studies.
Bioinformatics analysis
Datasets GSE158722, GSE102073, GSE28739 and GSE30161 in the Gene Expression Omnibus (GEO) database were analyzed to screen the upregulated pathways associated with EOC chemoresistance. Weighted Correlation Network Analysis 1.70-3 (WGCNA; a plug-in for R) was used for screening genes related to EOC chemoresistance and pathway enrichment analysis. DREAM and Gene Expression Profiling Interactive Analysis (GEPIA)2 databases were used to screen ABC molecules associated with PPARγ in EOC. Kaplan-Meier plotter database was used to analyze the relationship between PPARγ, ABCG2, angiogenin (ANG) and prognosis in EOC patients. STRING database and GeneCard database was used to predict the interaction between PPARγ and ABCG2. The corresponding websites for bioinformatics analysis are listed in Table SI.
Western blotting
Total proteins were extracted from OVCAR3 cells, OVCAR8 cells or murine OVCA tissues as previously described (29). Briefly, after harvesting the cells, RIPA (Jiangsu Kaiji Biotech. Co. Ltd.; cat. no. KGB5204) was used to lyse the cells. Protease inhibitors (1:1,000; Jiangsu Kaiji Biotech. Co. Ltd.; cat. no. KGB5101-100) were added to inhibit protease activity. A BCA assay kit (Jiangsu Kaiji Biotech. Co. Ltd.; cat. no. KGP902) was used to detect the protein concentration. Equal quantities of proteins (20 μg/lane) were separated by 10% SDS-PAGE, transferred onto PVDF membrane. The membranes were blocked for 2 h at 25°C with 3% BSA and incubated at 4°C overnight with specific primary antibodies against human PPARγ (1:1,000; Abcam; cat. no. ab59256), human ABCG2 (1:1,000; Cell Signaling Technology, Inc.; cat. no. 42078), GAPDH (1:1,000; Cell Signaling Technology, Inc.; cat. no. 2118), mouse ABCG2 (1:1,000; Cell Signaling Technology, Inc.; cat. no. 42078), mouse ABCB1 (a gene that encodes P-glycoprotein), 1:1,000; Cell Signaling Technology, Inc.; cat. no. 13978) and mouse β-actin (1:1,000; Cell Signaling Technology, Inc.; cat. no. 4967). The membranes were probed with the corresponding secondary antibodies [Goat Anti-Mouse IgG (H+L) HRP; 1:3,000; Affinity Biosciences; cat. no. S0002] at 25°C for 1.5 h and scanned using OdysseyCLx equipment (LI-COR Biosciences) for visualization of the bands. The density of the bands was measured using Odyssey software version 3.0 (LI-COR Biosciences).
Cell proliferation assay
Cell Counting Kit-8 (CCK-8 kit; Jiangsu Kaiji Biotech. Co. Ltd.; cat. no. KGA317) was used to detect cell proliferation. EOC cells were seeded in 96-well plates at 5×103 cells per well, cultured overnight at 37°C and treated as above. Subsequently, cell proliferation was assessed as previously described (30). Briefly, EOC cells were added with DMEM or 1640 medium containing 10% CCK-8 reaction solution and incubated at 37°C for 1.5 h. The absorbance was determined at 450 nm using a microplate reader (BioTek Instruments, Inc.).
Apoptosis assay
EOC cells were seeded into 6-well plates at 5×105 cells per well and cultured overnight at 37°C and treated with HATES, DDP/PTX, or other reagents for 48 h. The apoptosis of the cells was detected as previously described (30). Briefly, the apoptosis of the cells was determined using the FITC Annexin V Apoptosis Detection Kit with PI (Biolegend; cat. no. 640914) or PE Annexin V Apoptosis Detection Kit I (BD Biosciences; cat. no. 559763). After staining at 25°C for 30 min. The cells were collected using a flow cytometer (BD Biosciences) and 50,000 cells in each group were circled for analysis using FlowJo v10 software (FlowJo LLC). The cells that were Annexin V and 7-AAD/PI negative were considered viable; cells that were Annexin V positive and 7-AAD/PI negative were in early apoptosis; and cells that were both Annexin V and 7-AAD/PI positive were in late apoptosis or already dead. The apoptotic rate was the sum of the percentage of early and late apoptotic cells.
Flow cytometry analysis of ABCG2 expression
OVCAR3 cells were seeded into 6-well plates at 5×105 cells per well, cultured overnight at 37°C and treated with HATES for 48 h, with regular medium as a control. The cells were trypsinized and incubated with ABCG2 antibody (1:60; Abcam; cat. no. ab229193) at 25°C for 30 min, then incubated with Goat Anti-Rabbit IgG H&L (Alexa Fluor® 488; 1:2,000; Abcam; cat. no. ab150077) at 25°C for 30 min. Finally, the cell surface expression of ABCG2 was determined using a flow cytometer (BD Biosciences) and analyzed using FlowJo v10 software (FlowJo LLC).
Hoechst 33342 efflux assay
OVCAR3 cells were seeded into 24-well plates at a concentration of 1×104 cells per well and cultured overnight at 37°C. Then, the cells were treated with 0.4 g/ml HATES and 10 μM KO143 for 48 h. The culture media was discarded and 300 μl Hoechst 33342 (Cell Signaling Technology, Inc.) was added (final concentration of 1 μg/ml per well) and the plate was incubated overnight at 37°C (avoiding light during the entire process). After 1 h, the dye solution was discarded, 500 μl Hank's solution was added to each well and images were captured using a fluorescence microscope. The average fluorescence intensity was analyzed using ImageJ v2 (National Institutes of Health).
Dual luciferase assay
EOC cells were seeded into 24 well plates at a concentration of 2×105 cells per well. The ABCG2-promoter-Luc, GV238-v-luc (as the control) and β-gal plasmids (Beyotime Institute of Biotechnology) were transfected into cells using Lipofectamine® 3000 (Invitrogen; Thermo Fisher Scientific, Inc.). After transfection for 6 h, the medium was changed to regular medium. After incubation at 37°C for 48 h, the cells were lysed with 200 μl of lysis buffer. The cell lysates were then vortexed and centrifuged at 12,000 × g at 4°C for 2 min and 20 μl supernatant was added to 100 μl luciferase substrate to detect firefly luciferase activity (Promega Corporation; cat. no. E4030). The GloMax 20/20 Luminometer (Promega Corporation) was used to detect the ABCG2-promoter luciferase activity according to the supplier's recommendations. The values were normalized to the internal β-gal luciferase activities.
RNA sequencing
OVCAR3 cells were seeded into 6-well plates (5×105 cells per well). After culture at 37°C for 24 h, the cells were treated as follows: Control, HATES, DDP, DDP and HATES, PTX, or PTX and HATES at 37°C for 48 h. Subsequently, 1 ml TRIzol® (Thermo Fisher Scientific, Inc.) was added to lyse the cells and the samples were sent to Tiangen Biotech, Co., Ltd. for RNA sequencing and analysis.
Establishment of the DDP and PTX-resistant cell lines
DDP-resistant cells (OVCAR3-R/DDP) were obtained from parental cells by continuous treatment with gradually increasing doses of DDP (5-7.5 μM) for 6 months as previously described (31). Briefly, the cells were treated with 5 μM DDP for 24 h and the medium was changed to regular medium. When the cell confluence reached 80%, the cells were treated 5 μM DDP for 24 h again. The procedures were repeated until the cells could stably proliferate in the medium containing 5 μM DDP. The concentration was then increased to 7.5 μM and the procedures were repeated. The PTX-resistant cells were established in the same manner but with a dose range of 2-3 nM.
Small interfering (si)RNA transfection
Human PPARγ-siRNA and sham-siRNA (as a control) were synthesized and constructed by Shanghai GeneChem, Co., Ltd. The sequences of siRNA are listed in Table SII. OVCAR3 cells were seeded into 24-well plates (1×105 cells per well) and transfected with the plasmids using Lipofectamine® 3000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. After transfection for 12 h, the medium was changed to regular medium and the incubation continued for 48 h in the 37°C incubator. The cell lysates were collected and successful knockdown was confirmed using western blotting.
Immunohistochemistry (IHC) staining
A total of 16 HGSOC clinical specimens, including six chemo-sensitive (no recurrence 6 months after chemotherapy) and 10 chemoresistant (recurrence within 6 months of chemotherapy) samples, were collected from patients without preoperative therapy. The 4 μm pathological tissue section was dewaxed and antigen retrieval was conducted. The expressions of PPARγ and ABCG2 were detected with primary antibodies against PPARγ and ABCG2 (both 1:300; Abcam). The percentage of tissue positivity (positivity=strong positive percentage + positive percentage) was calculated.
Establishment of short hairpin (sh)PPARγ stable cell lines
PPARγ-RNAi lentivirus infection solution and negative control with shRNA vector GV344 (hU6-MCS-Ubiquitin-firefly_Luciferase-IRES-puromycin) were purchased from Shanghai GeneChem, Co., Ltd. The sequences of shRNA are listed in Table SIII. The cells were seeded in 6-well plates at 1×105/ml/well and incubated at 37°C and 5% CO2 for 24 h. Then the cells were infected with virus at 37°C for 12 h and the culture medium was replaced with regular medium. Puromycin (2 μg/ml) was used for selection and ≥5 clones were selected for cell expansion. The knockdown efficiency was confirmed by western blotting.
Animal experiments
All the mice used in the present study were purchased from Beijing Sibeifu Animal Company. The mice were kept in an environment of 40-60% relative humidity, room temperature and a 12-h light/dark cycle, and were allowed to acclimate to laboratory conditions for at ≥7 days before the experiment began. A total of 20 nude mice (16-18 g, 8-week-old, female, raised in specific pathogen-free conditions) were used to establish EOC animal models with OVCAR3 cells. First, OVCAR3-shNC cells (negative control) were intraperitoneally injected (5×106/200 μl/mouse) into 10 nude mice and the mice were randomly assigned to groups of shNC and shNC + DDP (n=5 per group). Second, OVCAR3-shPPARγ cells were intraperitoneally injected (5×106/200 μl/mouse) into another 10 nude mice and the mice were randomly assigned to groups of shPPARγ or shPPARγ + DDP (n=5 per group). The mice were intraperitoneally injected with DDP (1 mg/kg/w, beginning on the first day after tumor implantation). A total of 20 C57BL/6 mice (18-20 g, 8-week-old, female) were intraperitoneally injected with 5×106 ID8 cells/200 μl/mouse and randomly assigned to one of four groups (n=5 in each group): Control, GW9662(4 mg/kg), DDP (1 mg/kg/w) and DDP + GW9662. GW9662 (daily, started 1 day earlier than DDP) and DDP (started on the fourteenth day after tumor implantation). Treatments were intraperitoneally injected into the mice. Mice in the control group received an equivalent volume of PBS. Body weights were monitored daily. A total of 4 (nude mice) or 5 (C57BL/6 mice) weeks after tumor implantation, when the control mice showed a maximum 30% increase in their body weight, all mice were deeply anesthetized by exposure to 2% isoflurane for 10 min and then sacrificed by cervical dislocation for analysis. The abdominal tumor nodules were observed, counted and weighed. The livers of nude mice were fixed using 4% formalin at 4°C for 48 h for histological analysis as described previously (32).
Cytokine array analysis
After the HATES samples from EOC patients were prepared, two HATES samples were randomly selected and sent to Shanghai Aksomics. Biotech, Co., Ltd. for cytokine array analysis.
Statistical analysis
Data are presented as the mean ± standard deviation (SD) and were analyzed using GraphPad Prism version 8.0 (GraphPad Software, Inc.). Multiple comparisons were performed using a one-way ANOVA with the Tukey post hoc test. An unpaired t-test was used to compare the difference between two independent samples. P<0.05 was considered to indicate a statistically significant difference.
Results
PPARγ and ABCG2 are involved in ARM-related chemoresistance in EOC
To explore the possible adipocyte-rich associated mechanisms in EOC, first, HATES from peritumoral adipose tissue of EOC patients was applied to mimic ARM in vitro. HATES was co-cultured with the human HGSOC cell line, OVCAR3 cells and cells were treated with DDP or PTX. Additionally, mouse ID8 EOC cells were co-cultured with mouse adipose tissue extracts (MATES) from the peritumoral adipose tissue of an EOC C57BL/6 mouse model and subsequently treated with DDP or PTX. At the prepared concentrations, HATES or MATES did not in themselves influence the morphology of cancer cells (Fig. 1Aa), proliferation (Fig. 1Ab), or apoptosis (Fig. 1Ac). Under the same conditions, HATES or MATES culture significantly promoted cell proliferation and decreased the apoptosis rate when cells were treated with either DDP or PTX. MATES co-culture reduced the apoptosis rate of ID8 cells from 34.6% (DDP treatment) and 26.86% (PTX treatment) to 6.42 and 5.49%, respectively (Fig. 1Ac). These preliminary results suggested that HATES/MATES promote chemoresistance of human/mouse EOC cell lines.
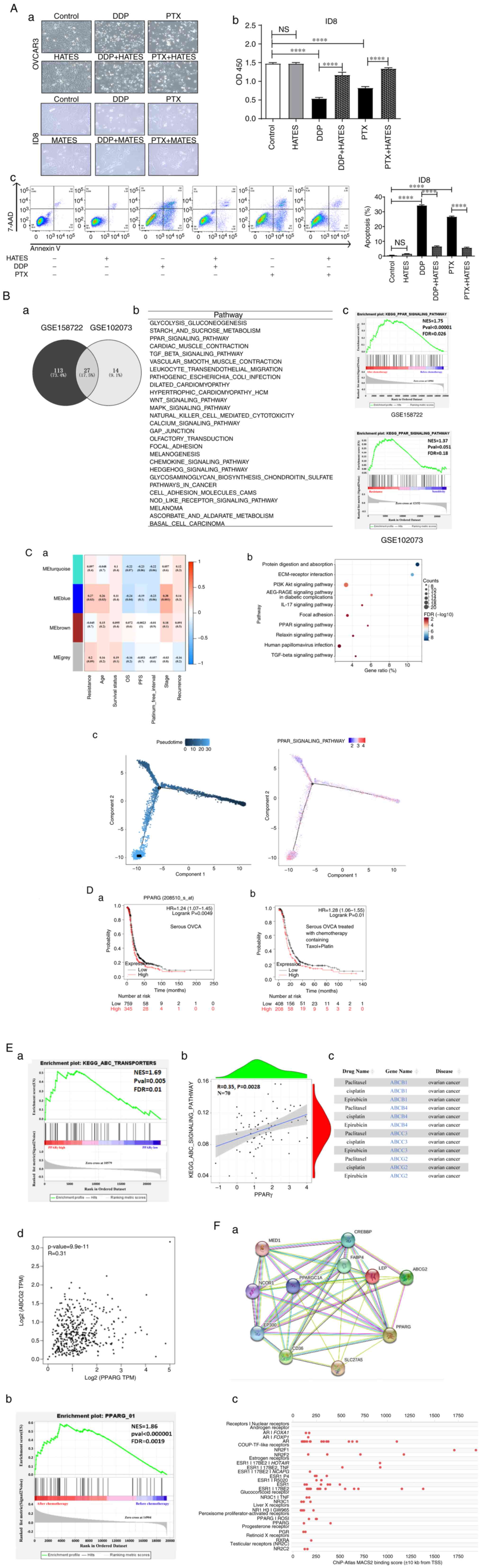
Bioinformatics analysis was used to investigate the possible ARM that was involved in the above effects with regard to chemoresistance. First, GEO was used to screen out 27 common upregulated pathways in the datasets GSE158722 (EOC cells) and GSE102073 (EOC patients) datasets, both of which contain chemotherapy information (Fig. 1Ba and Bb). It was found that the PPAR signaling pathway, which is closely related to lipid metabolism, was significantly enriched. Fig. 1Bc shows the enrichment analysis of the PPAR signaling pathway in dataset GSE158722 (FDR <0.05) and dataset GSE102073 (FDR <0.25). To verify the correlation between the PPAR signaling pathway and EOC resistance, WGCNA was used to further analyze 70 patients who received chemotherapy in the GSE102073 dataset (Fig. 1Ca). Fig. 1Cb shows the top 10 signaling pathways that were significantly positively associated with chemoresistance based on KEGG enrichment analysis, which included the PPAR signaling pathway (FDR=0.008). A pseudo-timing analysis of the PPAR signaling was also performed in EOC cells after chemotherapy in the GSE158722 dataset and it was found that PPAR signaling in EOC cells gradually increased with the pseudo-timing (Fig. 1Cc). The PPAR family consists of three subtypes including PPARα, PPARγ and PPARβ/δ (33). To determine which PPAR molecule is primarily involved in chemoresistance in OVCA, Kaplan-Meier analysis was performed. PPARα and PPARβ/δ did not show a consistent statistical association (data not shown), whereas PPARγ mRNA expression was significantly associated with a worse progression-free survival (PFS) in patients with serous OVCA (Fig. 1Da; n=1,104; P<0.005) and serous OVCA who received chemotherapy consisting of Taxol (i.e. PTX) and Platin (Fig. 1Db; n=616; P=0.01). Moreover, in patients with serous OVCA (Stage, 3+4; grade, 3), that is HGSOC, receiving Platin and/or Taxol treatments, PPARγ was significantly correlated with a worse PFS (Fig. S1A). Thus, the bioinformatics data indicated that PPARγ played a crucial role in the mechanism of ARM-related chemoresistance in OVCA.
To study the mechanism by which PPARγ promotes OVCA resistance, the GSE102073 dataset was used and GSEA was performed to screen PPARγ-related downstream molecules associated with chemoresistance. Fig. 1Ea shows that the ABC signaling pathway was significantly enriched when PPARγ was highly expressed (FDR=0.01). There was a significant positive correlation between PPARγ and the ABC signaling pathway (R=0.35; P=0.0028; Fig. 1Eb). The DREAM database was used and it screened out ABCB1, ABCB4, ABCC3 and ABCG2 in relation to OVCA chemoresistance (Fig. 1Ec). Next, GEPIA2 was used for further analysis and it was found that PPARγ was significantly positively correlated with ABCB1 (R=0.15; P=0.0013; Fig. S1B) and ABCG2 (R=0.31; P=9.9×10-11; Fig. 1Ed). Compared with ABCB1, ABCG2 was more significantly correlated with PPARγ in OVCA. To confirm the correlation between PPARγ and ABCG2, the STRING database and GeneCards data were used for further analysis. The interactions between PPARγ and ABCG2 proteins are shown in Fig. 1Fa. Additionally, PPARγ also interacted with various proteins involved in tumor lipid metabolism, such as CD36, fatty acid binding protein 4 (FABP4) and leptin, indicating that there were plenty of molecules that can interact with PPARγ in ARM. GSEA was performed for transcription factors of ABCG2 in the GSE158722 dataset and the results revealed that PPARγ was significantly enriched in EOC cells following chemotherapy (FDR=0.0019; Fig. 1Fb). The prediction results of Chip-SEQ data showed that PPARγ was a transcription factor of ABCG2 (FDR=1E-05; Fig. 1Fc). These data suggested that PPARγ was a transcription factor of ABCG2 in EOC. These results provide evidence for the involvement of PPARγ in chemoresistance probably via its upregulation of ABCG2 in EOC.
HATES enhances ABCG2 expression via upregulation of PPARγ in EOC cells
To verify the mechanism by which ARM influences ABCG2 via upregulation of PPARγ, luciferase reporter assays in OVCAR3 cells and OVCAR8 cells were performed. In both cell lines, the PPARγ antagonist, GW9662, inhibited the transcription activity of ABCG2; whereas the PPARγ agonist, Trog, enhanced the transcription activity of ABCG2 (Fig. 2Aa). Treatment with HATES increased the transcription of ABCG2, which was decreased by GW9662 and increased by Trog (Fig. 2Ab), suggesting that HATES promotes the transcriptional activity of ABCG2 via PPARγ in the two cell lines. Furthermore, treatment of HATES increased protein expression of PPARγ and ABCG2, whereas GW9662 decreased the expression of ABCG2, which was then partially reversed by the addition of HATES (Fig. 2B).
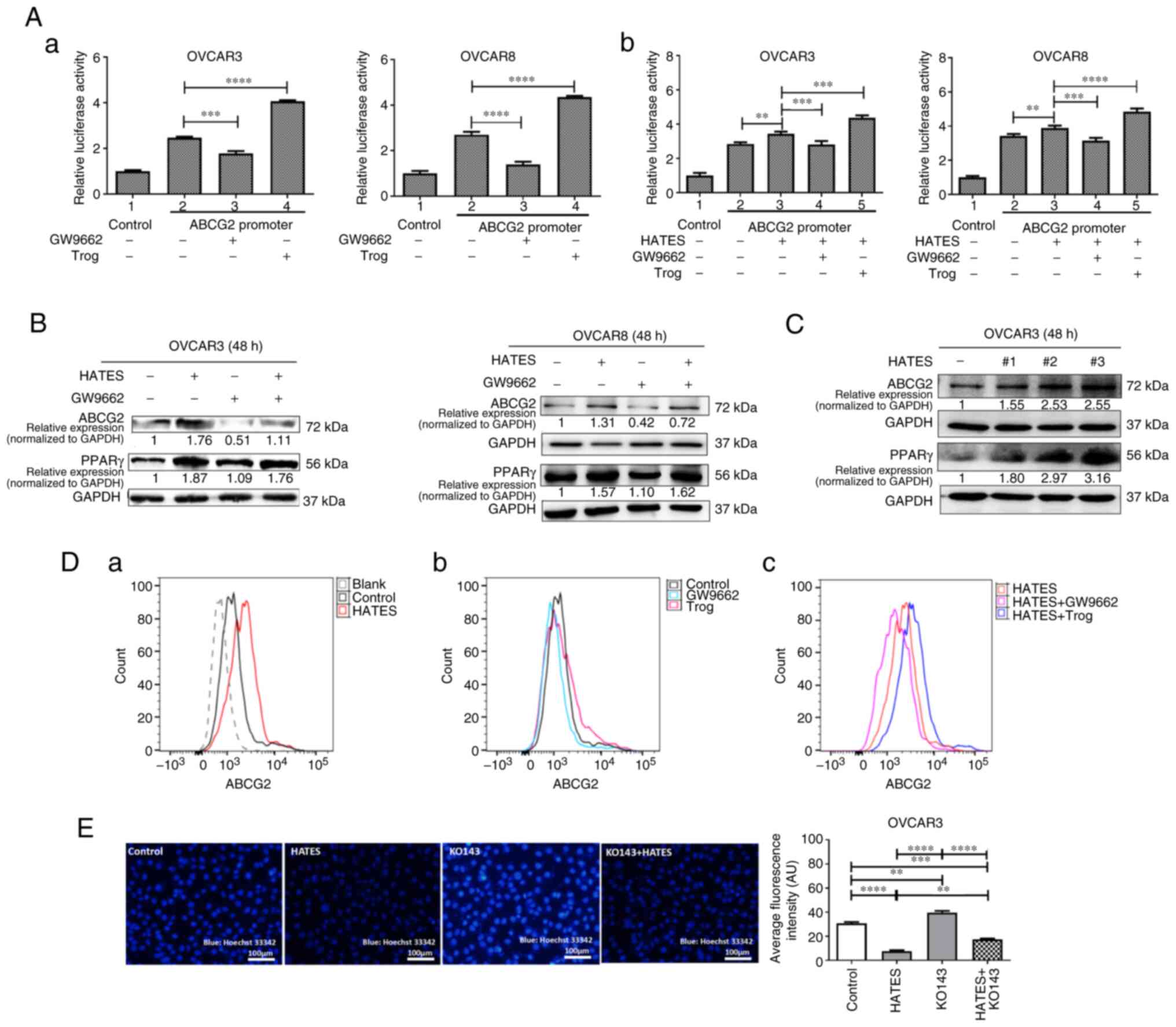
To determine whether the effects of HATES derived from different patients were specific or universal, three HATES samples from peritumoral adipose tissue of three EOC patients were used to treat OVCAR3 cells for the following mechanistic experiments. All three HATES samples significantly increased the expression of PPARγ and ABCG2 in OVCAR3 cells compared with the control (Fig. 2C). To confirm the effects of HATES on the expression of ABCG2, HATES sample #2 in Fig. 2C was randomly selected to treat OVCAR3 cells together with Trog and GW9662. Treatment with HATES significantly increased the expression of ABCG2 on the surface of OVCAR3 cells compared with the control (Fig. 2Da). Treatment with GW9662 alone decreased the expression of ABCG2 (Fig. 2Db) and the increased expression of ABCG2 induced by HATES was attenuated by the addition of GW9662 (Fig. 2Dc). By contrast, treatment with Trog alone increased the expression of ABCG2 (Fig. 2Db). The increased expression of ABCG2 induced by HATES was further increased by the addition of Trog (Fig. 2Dc).
ABCG2 contributes to MDR by enhancing the extrusion of drugs when it is overexpressed in cancer cells. Thus, HATES sample #3 was used to determine whether ARM influences the extrusion function of ABCG2. Compared with the control group, HATES markedly increased the efflux of Hoechst 33342 and reduced the intracellular fluorescence intensity in OVCAR3 cells (Fig. 2E, P<0.0001), indicating that HATES improved the extrusion function of ABCG2. KO143 blocked the function of ABCG2, decreasing Hoechst 33342 extrusion and subsequently leading to an increase in intracellular fluorescence intensity compared with the control (P<0.01). Treatment with HATES partially increased the reduced ABCG2 function induced by KO143 and recovered the intracellular fluorescence intensity (P<0.0001). Together, these results indicate that ARM not only enhanced ABCG2 expression via upregulation of PPARγ but also facilitated the extrusion function of ABCG2 in EOC cells.
HATES promotes chemoresistance by increasing ABCG2 expression via upregulation of PPARγ
To determine whether the effects of ARM on PPARγ and ABCG2 contribute to chemoresistance, the transcriptome of OVCAR3 cells after treatment with DDP or PTX with or without HATES was determined by RNA transcriptome sequencing. The mRNA levels of PPARγ and ABCG2 were both increased after treatment with HATES when the cells were treated with or without chemotherapy (Fig. 3A), suggesting that HATES affected chemoresistance by regulating PPARγ and ABCG2 in OVCAR3 cells. HATES also increased the protein expression levels of PPARγ and ABCG2 when the cells were treated with DDP and PTX (Fig. 3B). Since the present study primarily mainly focused on the influence of ARM-educated PPARγ and ABCG2 on chemoresistance, Trog, GW9662 and KO143 were all applied as pretreatments of 2 h to avoid affecting cell proliferation or apoptosis. Next, cells were treated with HATES for 48 h based on the data in Fig. S2. Neither separate nor combined treatments of HATES with Trog, GW9662, or KO143 affected the proliferation of both OVCAR3 and OVCAR8 cells (Figs. 3Ca and S2). Compared with the HATES + chemotherapy treatment groups, the addition of GW9662 and KO143 significantly decreased cell proliferation (Figs. 3Ca and S2) and increased the apoptosis of OVCAR3 cells (Fig. 3Cb). These results indicated that HATES promotes chemoresistance via the PPARγ/ABCG2 pathway.
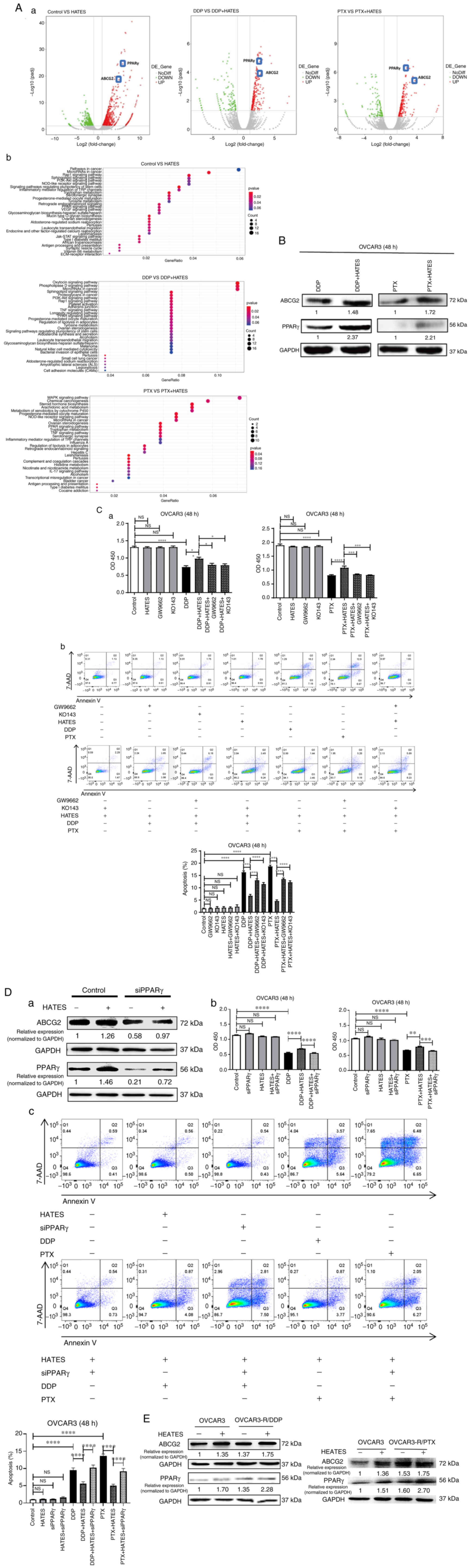
To examine whether HATES promoted chemoresistance via upregulation of PPARγ/ABCG2 endogenously, siPPARγ was transiently transfected in OVCAR3 cells. Knockdown of PPARγ not only decreased the expression of ABCG2 but also attenuated the increase in protein expression of both PPARγ and ABCG2 induced by HATES treatment (Fig. 3Da). As shown in Fig. 3Db, the increase in cell proliferation induced by HATES could also be partially inhibited by the knockdown of PPARγ when treated with either DDP (P<0.0001) or PTX (P<0.001). HATES significantly decreased the apoptosis rate of OVCAR3 cells treated with DDP or PTX, whereas PPARγ silencing partially reversed the effects of HATES on apoptosis (both P<0.0001, Fig. 3Dc).
DDP-resistant (OVCAR3-R/DDP) and PTX-resistant OVCAR3 cells (OVCAR3-R/PTX) were also established to determine whether HATES exerted effects on chemoresistant cancer cells. Compared with the parental cells, both chemoresistant OVCAR3 cells exhibited higher protein expression levels of PPARγ and ABCG2. Additionally, HATES further increased the protein expression of PPARγ and ABCG2 in both chemoresistant cells (Fig. 3E). These results confirmed that HATES promoted chemoresistance by increasing ABCG2 expression via PPARγ in OVCAR3 cells.
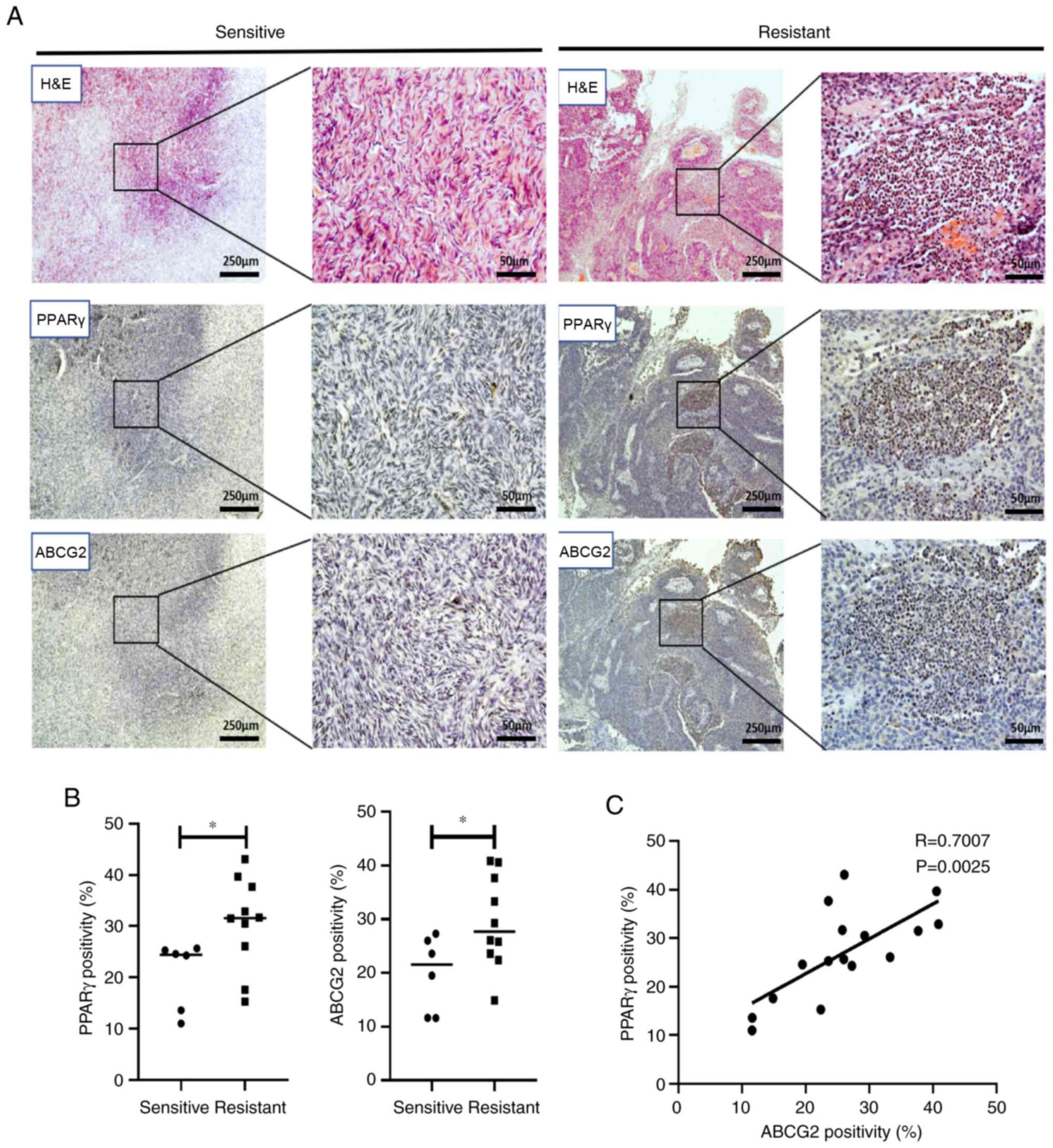
Expression of PPARγ and ABCG2 are associated with chemoresistance in EOC clinical specimens
To determine whether ARM contributes to chemoresistance via PPARγ and ABCG2 in patients, chemosensitive and chemoresistant EOC clinical specimens were collected (Fig. 4A). The expression of PPARγ and ABCG2 in chemoresistant EOC specimens was significantly higher than those in chemosensitive specimens (both P<0.05; Fig. 4B) and ABCG2 was positively correlated with PPARγ expression (R=0.70; P=0.0025, Fig. 4C), suggesting that the expression of PPARγ and ABCG2 was related to chemoresistance in EOC clinical specimens. The information on the stages and metastatic status of patients is shown in Table SIV.
Intervention of PPARγ affects the efficacy of chemotherapy in an EOC mouse model
To further evaluate the effects of ARM on chemoresistance via the PPARγ/ABCG2 pathway, an EOC mouse model was established and PPARγ function was inhibited both endogenously and exogenously. Since OVCAR3 cells are DDP-resistant (34), DDP was used for chemotherapy in the EOC mouse model.
First, for the endogenous intervention, immunodeficient BALB/C EOC mouse models were established by intraperitoneal injection of PPARγ knockdown OVCAR3 cells (Fig. 5Aa) and then the mice were treated with DDP. Mice in both OVCAR3-shNC and OVCAR3-shPPARγ groups developed ascites (the former showed considerably more ascites than the latter), whereas mice in the other two DDP-treated groups did not (Fig. 5Ab). Therefore, the tumor nodules (pointed by red arrows) on the external wall of the intestine in the two DDP-treated groups were compared. Compared with the DDP-treated OVCAR3-shNC group, mice in the DDP-treated OVCAR3-shPPARγ group demonstrated fewer tumor nodules and they were lower in weight (both P<0.05; Fig. 5Ac). OVCAR3 cells were more likely inclined to develop liver metastases than to form malignant ascites (35). Therefore, the status of liver metastases was examined using hematoxylin and eosin staining. The livers of mice in the OVCAR3-shPPARγ group showed less destroyed hepatic lobule structures, less disordered liver cells, fewer inflammatory cells than those in the OVCAR3-shNC group and the two DDP-treated groups did not exhibit any liver inflammation or disorder in the liver of mice (Fig. 5Ad). No cancer cells were detected in the livers of the four groups. The weights of the mice were monitored every other day (Fig. S3A).
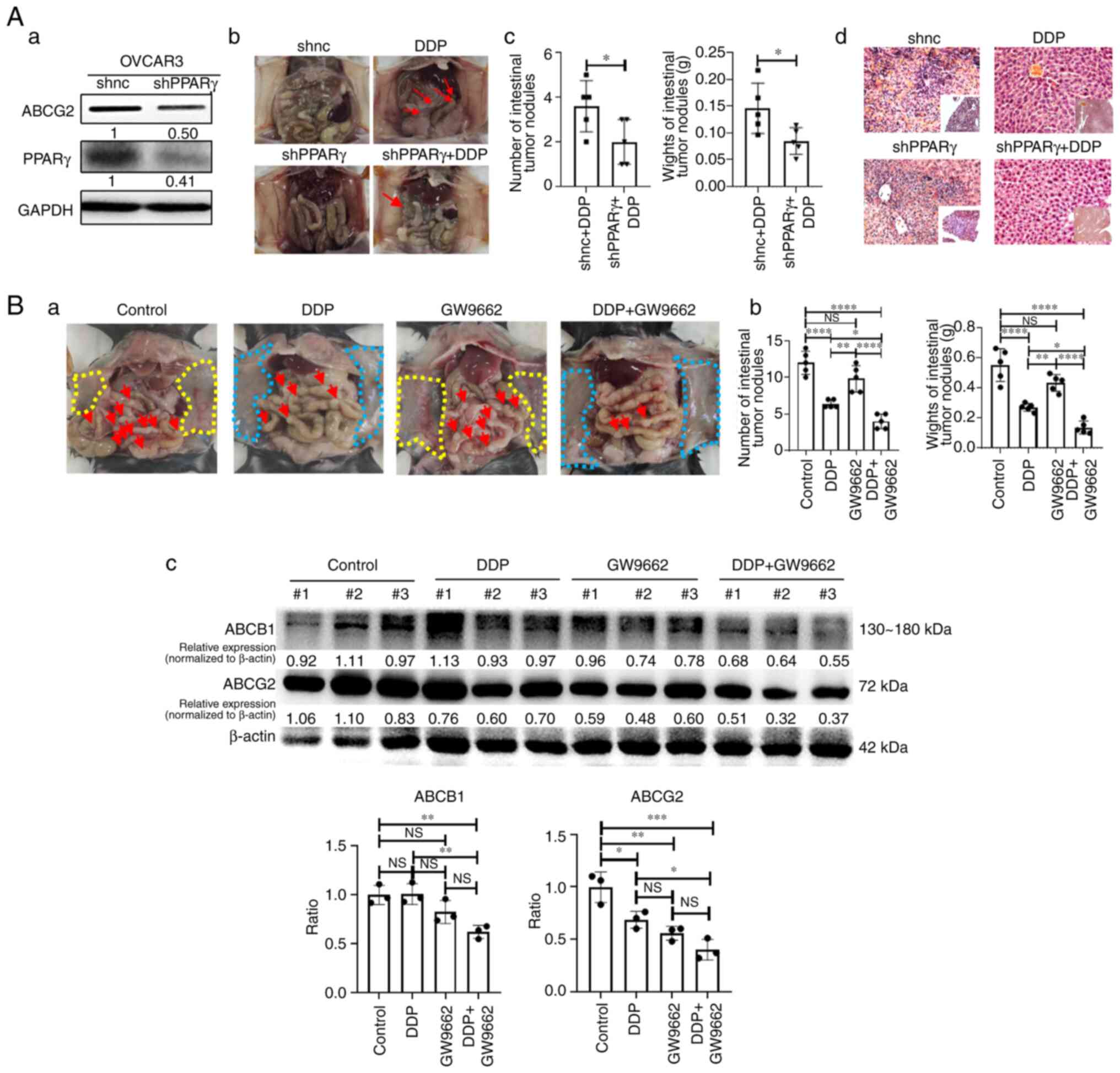
For the exogenous intervention, an immunocompetent syngeneic EOC mouse model was established by intraperitoneal injection of ID8 cells into C57BL/6 mice. GW9662, a PPARγ antagonist, was used as an exogenous adjuvant combined with DDP to determine the effects in the mouse model. Mice in the two DDP-treated groups (DDP and DDP + GW9662; Fig. 5Ba) demonstrated smooth and clear abdominal walls (area circled by the blue dotted line) and notably fewer tumor nodules on the external wall of the intestinal tracts (red arrows) than those of the control and GW9662 groups, both of which showed numerous tumor nodules on the abdominal wall (area circled by the yellow dotted line; Fig. 5Ba). Additionally, the combination of DDP and GW9662 resulted in fewer tumor nodules and lower-weight tumors compared with the DDP group (P<0.05; Fig. 5Bb). Mice in the GW9662 group presented a trend of fewer tumor nodules than the control group; however, there was no significant difference between the two groups. Tumor samples were randomly selected from three mice in each group for western blotting and the protein expression levels of ABCG2 in the combined treatment group confirmed the inhibitory effect of GW9662 and its expression was lower than that in the DDP group (Fig. 5Bc; P<0.05). Additionally, the expression of ABCG2 and ABCB1, with which PPARγ was significantly positively correlated in Fig. S1B, was also detected. The protein expression levels of ABCB1 in the combined group were also lower compared with the DDP group as well (Fig. 5Bc; P<0.01). Although the protein expression levels of ABCG2 and ABCB1 in the GW9662 group did not demonstrate a significant decrease compared with that of the control group, they showed a trend of reduction in the protein expression levels. The weights of the mice were monitored every other day (Fig. S3B).
Together, these results showed that in the mouse models, both endogenous and exogenous interventions of PPARγ may improve the chemotherapeutic efficacy and attenuate the chemoresistance to a certain extent.
ANG and oleic acid (OA) play key roles in HATES to upregulate PPARγ
In ARM, adipose tissue secretes various factors such as fatty acids, lipids and adipokines. Two HATES samples were randomly selected (#2 and #3 in Fig. 2C) from EOC patients and cytokine array analysis was performed (Fig. 6Aa). Hierarchical clustering analysis showed the presence of numerous adipokines that were simultaneously expressed in both HATES samples, such as ANG, TIMP-2 and leptin, of which ANG and TIMP-2 were both highly expressed in both HATES (Fig. 6Aa and Ab). Bioinformatics analysis was then performed and it was found that ANG was correlated with PPARγ in the EOC GSE28739 dataset (Fig. 6Ba; R=0.8688; P<0.0001) and in the HGSOC GSE30161 dataset (Fig. 6Bb; R=0.5164; P<0.05). The GSE28739 dataset contains 30 drug-resistant EOC cases and the GSE30161 dataset includes 18 patients with carboplatin/PTX (partial response) for stage III and IV EOC. Kaplan-Meier analysis showed that ANG mRNA expression was significantly associated with a worse PFS in patients with serous OVCA treated with chemotherapy containing Platin (Stage, 3+4; Grade, 3; P<0.05; Fig. 6C). No significant data for TIMP-2 was available in the EOC database, although TIMP-2 was highly expressed in the HATES. These data suggested that ANG is a major adipokine in HATES that exerts effects on PPARγ and subsequently influences chemoresistance.
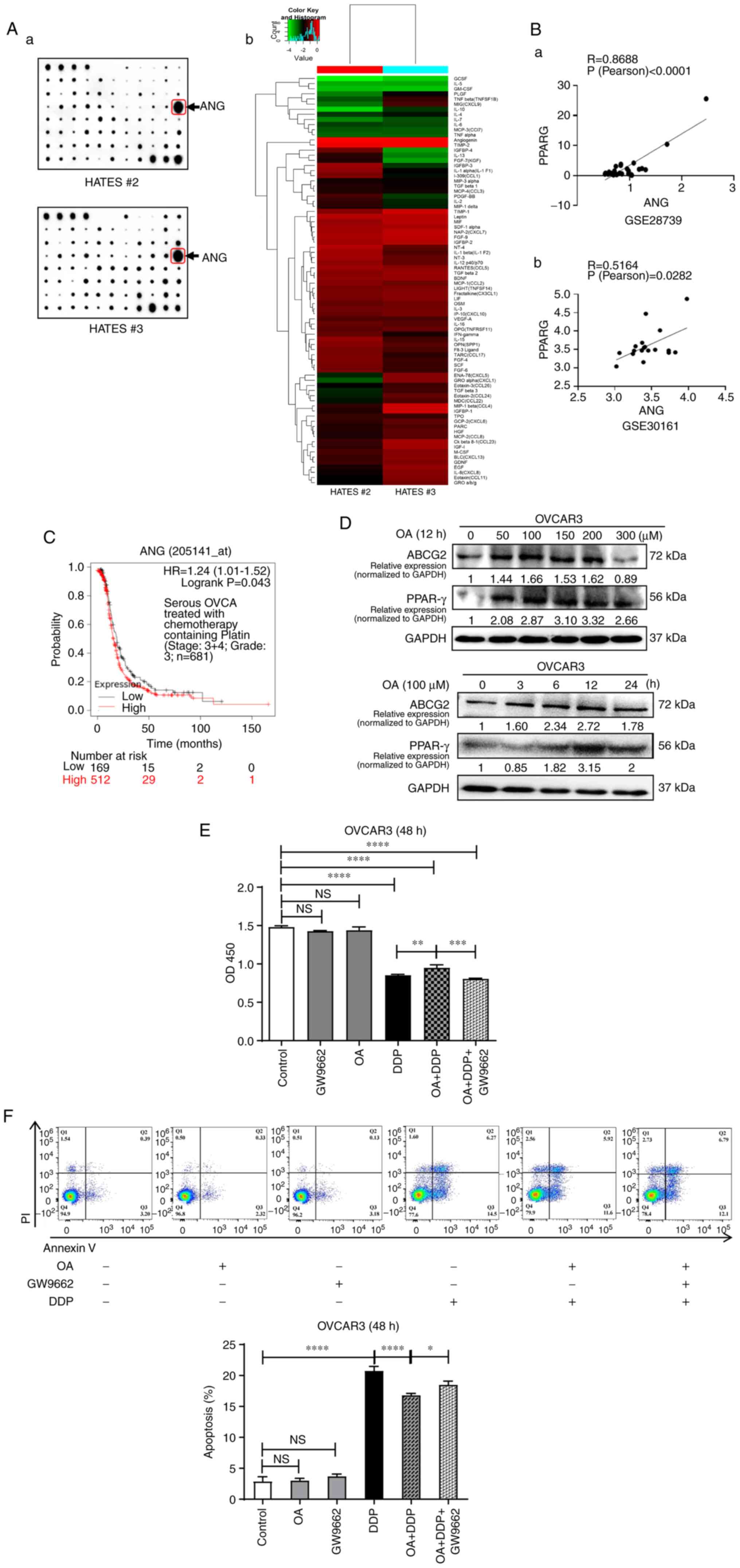
Saturated fatty acids can induce endoplasmic reticulum stress and cause apoptosis of cells (36-38). Hence, the present study used OA, one of the most common free fatty acids present in serum and a monounsaturated fatty acid (MUFA), to determine the effects on PPARγ/ABCG2. The protein expression of PPARγ and ABCG2 were increased in a time- and dose-dependent manner when treated with OA (Fig. 6D). OA treatment increased the proliferation of OVCAR3 cells treated with DDP, whereas GW9662 partially attenuated the effects of OA treatment (Fig. 6E). Furthermore, OA treatment decreased the apoptosis rate of OVCAR3 cells treated with DDP, which could be partially recovered by the addition of GW9662 (Fig. 6F). However, the effects of OA on apoptosis were not as potent as the effects of HATES (Fig. 3Dc), indicating that OA played a partial role in promoting chemoresistance in HATES.
Discussion
The mechanisms underlying chemoresistance are complex and include various aspects, among which increased drug efflux mediated by drug pumps is a common feature of chemoresistant cells (39). Therefore, current strategies primarily focus on repositioning or repurposing of chemotherapeutic agents to re-sensitize MDR cancer cells (40-43). ABC transporters are key proteins that extrude drugs out of cancer cells (44,45). Although numerous ABCG2 inhibitors have been developed, there have been no successful clinical trials published in the re-sensitization of ABCG2-mediated chemoresistance as of yet (46). Additionally, the adverse drug interactions and side effects of these ABCG2 inhibitors (47,48) and the influence of the TME, such as ARM, may be another concern affecting the function of ABCG2 transporters and subsequently influencing the application of the inhibitors. It has thus been suggested that any therapeutic strategy should be more context-dependent (7). The present study is the first study, to the best of the authors' knowledge, targeting an ARM-educated molecule, PPARγ, to upregulate ABCG2 and re-sensitize chemoresistant cancer cells, highlighting a potential method for improving the efficacy of chemotherapy through a mechanistic understanding of the TME.
Adipose tissue is composed of various cells, such as adipocytes, fibroblasts, immune cells and endothelial cells, amongst other components. These cells secrete fatty acids, lipids and adipokines including adipocytokines, which contribute to chemoresistance through different mechanisms, including metabolic reprogramming and inflammatory changes (49-52). The roles of the most common adipokines in health and disease (53-56) and especially in chemoresistance (49,57), have been reviewed previously. For example, adiponectin, IL-6 and IL-8 have been reported to be the most abundantly present adipokines secreted by omental adipocytes (56). These adipokines play an important role in the TME of OVCA and thus affect cancer development and metastasis (56). IL-6 can also enhance MDR of breast cancer cells via ABCG2 upregulation (58). Leptin promotes chemoresistance through various mechanisms in different types of cancer (14). For example, leptin can upregulate Notch/RBP-Jk signaling, which is associated with cancer and chemoresistance and inhibition of leptin significantly reduces the expression of ABCB1, which is highly specific for PTX efflux in cancer cells (59).
The predominant influence of ARM affecting chemoresistance is through the combined effect of fatty acids and certain major adipokines, which was confirmed by cytokine array analysis of HATES samples in the present study. The heterogeneity of the TME in the patients intensified the assessment of the complexity of this combined effect. Hence, it is a significant challenge to identify which and how many adipokines are primarily involved in collaborating and regulating the PPARγ/ABCG2 pathway. The most predominant adipokine in HATES was ANG, which contributed to angiogenesis and tumor progression (55,60) and was notably correlated with PPARγ in EOC patients receiving chemotherapy based on data from two GEO datasets. HATES is considered an excellent cell-free angiogenesis-inducing agent for vascularization (61,62). A study in colorectal cancer confirmed the angiogenic function of peritumoral adipose tissue as well (63). Antiangiogenic drugs have been implicated in the treatment of different types of cancer (64-66). Preclinical results have shown the association between antiangiogenic chemoresistance and ARM (49,67) and the relatively high proportion of ANG in HATES in the present study may explain the corresponding mechanism to a certain extent. Studies in breast cancer have reported that an altered adipokine profile provides a potential mechanism contributing to chemoresistance, in which IL-6, TNF-α and leptin function as potential mediators (68,69). Yang et al (70) negated the involvement of leptin, adiponectin, PGE2 and lipoxin A4 in adipocyte-induced chemoresistance in OVCA. They applied conditioned medium from adipocytes and found that adipocytes induced chemoresistance in OVCA cells via arachidonic acid (omega-6 polyunsaturated fatty acid; PUFA) via activation of Akt (70). PUFAs and their metabolites are agonists of PPARγ (71) and even small amounts of PUFAs can induce chemoresistance (72,73). PUFAs from astrocytes have been shown to function as an activator of PPARγ to facilitate brain metastasis (74). Hence, PUFAs may be another regulator in HATES to upregulate PPARγ, which needs to be further investigated. In addition to adipokines, it was also determined that OA, an MUFA and representative fatty acid in HATES, influenced the PPARγ/ABCG2 pathway and confirmed its effects on chemoresistance. The effects of OA on apoptosis of OVCAR3 cells were not as potent as the effects of HATES. This difference further showed that other factors, for example, adipokines and PUFAs as aforementioned, contribute to the effects of HATES as well. OA has been reported to promote a malignant phenotype and contribute to chemoresistance in prostate cancer (75), in agreement with the findings of the present study.
It is well established that adipocytes play crucial roles in tumor progression in various adipocyte-rich associated cancers. For example, adipocytes supply energy to cancer cells as fatty acids, promote growth and metastasis in OVCA (7) and drive melanoma progression via fatty acid transport proteins (FATPs) (76). In the present in vitro experiments, considering that the primary focus was on the effects of ARM on chemoresistance, it was necessary to exclude the influence of ARM itself on the proliferation and apoptosis of cancer cells. Therefore, the experimental conditions were assessed to ensure HATES and OA did not cause any statistically significant changes in the proliferation or apoptosis of cancer cells, that is, HATES was applied with at least half of the in vivo concentration; cells were pretreated with Trog, GW9662 and KO143 for 2 h and cells were treated with 100 μM OA and this did not alter the proliferation of cancer cells, in agreement with a study by Zhang et al (77). Additionally, the half-life of GW9662 in cell culture is only ~2 h (78), which should be noted for in vitro and in vivo studies.
PPARγ exerts anti-tumor or pro-tumor effects by affecting multiple pathways (23,24,79,80). In previous studies on OVCA, there were contradictory opinions regarding the roles of PPARγ between clinical observations and laboratory research. Zhang et al (81) found that PPARγ was involved in the initiation and progression of OVCA and was significantly increased in high-grade OVCA (grade 3) compared with normal ovaries and benign tumors. However, several in vitro studies on OVCA have reported that different PPARγ agonists or ligands inhibit the proliferation of OVCA cell lines (82-85). In vivo studies also showed that ciglitazone, a PPARγ agonist, inhibited the growth of OVCA (83), especially when combined with DDP (86). The results of the present study may resolve this contradiction, since in previous studies, PPARγ agonists were applied alone without considering any TME factors, especially ARM. In the present in vivo experiments, both immunodeficiency and immunocompetent syngeneic mouse models were used to observe the effects of PPARγ intervention, thus taking into consideration the microenvironmental effects of not only ARM but also immune factors. It was also found that the treatment with GW9662 alone or knockdown of PPARγ alone demonstrated a tendency to inhibit tumor growth, partially supporting Zhang et al (81) in clinical observations, although the difference was not significant compared with the control.
GW9662 exerts antitumor activity in different preclinical models (87). In addition to the present study, GW9662 can also increase anti-PD-L1/PD-1 antibody efficacy and improve the efficacy of immunotherapy (88). Therefore, GW9662 may not only be a promising adjuvant agent for chemotherapy to reverse chemoresistance but also be a beneficial candidate for chemoimmunotherapy. Further studies are required to optimize the timing, concentration and duration of treatment with PPARγ antagonists to decrease the dosage of chemotherapeutic drugs and consequently reduce the undesirable side effects of chemotherapy.
The present study primarily focused on in vitro studies and in vivo experiments. It should be noted that there are limitations to this study since the ARM of OVCA in humans is considerably more complicated than that in mice and in vitro settings. There may be a huge difference in the fatty acid metabolism and adipokine profile between humans and mice. The heterogeneity of human OVCA should also be considered. Therefore, further exploration is required before PPARγ antagonists can be used in a clinical setting.
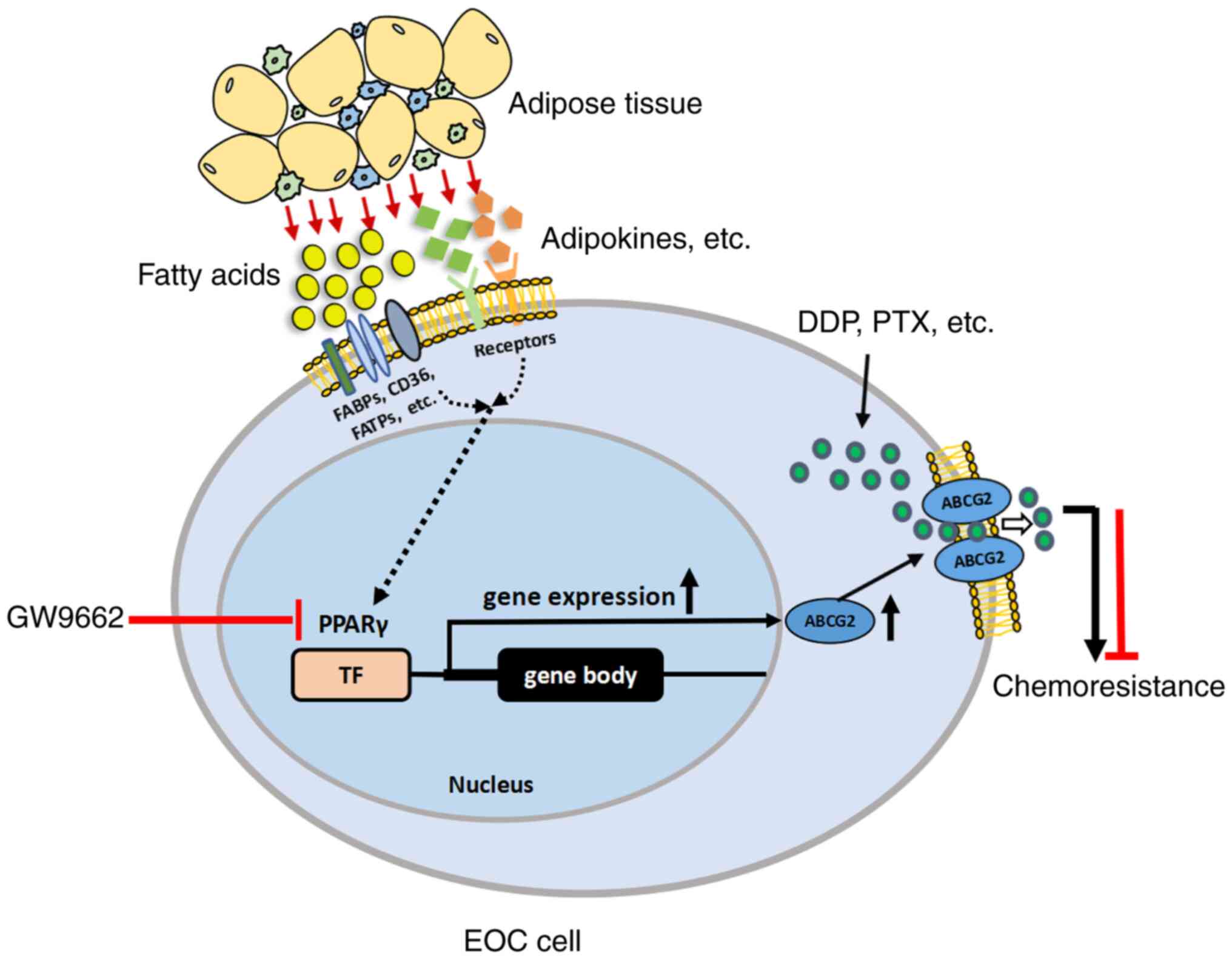
In summary, adipose tissues secrete various factors, including fatty acids, lipids and adipokines. Fatty acids are transported into EOC cells by fatty acid binding proteins such as FABPs, CD36 and FATPs. Adipokines exert their effects by binding with their respective receptors. ARM provides fatty acids and adipokines for cancer cells, resulting in the enhancement of PPARγ, subsequently leading to an increase in its downstream gene ABCG2, which pumps drugs out of cancer cells and causes MDR. Therefore, ARM promotes chemoresistance by increasing ABCG2 expression via upregulation of PPARγ (Fig. 7). The present study showed that antagonism/inhibition of PPARγ could attenuate the chemoresistance of EOC and may serve as a novel therapeutic adjuvant to chemotherapy. In the present study, the TME was considered with a focus on ARM-educated molecules to explore the re-sensitization of chemoresistant cancer cells. It was hypothesized that ARM may exert its effects through a common and non-exclusive mechanism to facilitate multi-chemoresistance in adipocyte-rich associated cancers, which provides a clue for identifying a novel therapeutic target related to ARM to improve the efficacy of chemotherapy or combined therapies.
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
SC was responsible for formal analysis, investigation, validation and editing. ZL was responsible for investigation, methodology and validation. HW was responsible for investigation, methodology and validation. BW was responsible for resources and investigation. YO was responsible for methodology and validation. JL was responsible for formal analysis, methodology and investigation. XZ was responsible for formal analysis, methodology and validation. HZ was responsible for resources, methodology and validation. XL was responsible for resources, methodology and validation. XF was responsible for resources, methodology and validation. YL was responsible for formal analysis, methodology and validation. YS was responsible for investigation, methodology and validation. HZ was responsible for resources and investigation. BX was responsible for methodology and validation. CY was responsible for conceptualization, formal analysis, investigation and writing the original draft. WD was responsible for conceptualization, funding acquisition, project administration, supervision, writing-review and editing. All authors read and approved the final manuscript. WD and CY confirm the authenticity of all the raw data.
Ethics approval and consent to participate
All procedures were in accordance with the ethical standards of Tianjin Central Hospital of Gynecology Obstetrics (approval number 2017KY009) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Written informed consent was obtained from all the participants prior to participation in the study. All animal experiments were performed following the protocol approved by the Animal Ethical and Welfare Committee (AEWC) of Tianjin Medical University (protocol number SYXK 2019-0004).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from National Natural Science Foundation of China (grant no. 82273340 to W. Deng) and Beijing-Tianjin-Hebei Basic Research Cooperation Project (grant no. 20JCZXJC00140 to W. Deng).
References
|
Miller KD, Nogueira L, Devasia T, Mariotto AB, Yabroff KR, Jemal A, Kramer J and Siegel RL: Cancer treatment and survivorship statistics, 2022. CA Cancer J Clin. 72:409–436. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Raghavan S, Winter PS, Navia AW, Williams HL, DenAdel A, Lowder KE, Galvez-Reyes J, Kalekar RL, Mulugeta N, Kapner KS, et al: Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell. 184:6119–6137.e26. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Lin F, Li X and Wang X, Sun H, Wang Z and Wang X: Stanniocalcin 1 promotes metastasis, lipid metabolism and cisplatin chemoresistance via the FOXC2/ITGB6 signaling axis in ovarian cancer. J Exp Clin Cancer Res. 41:1292022. View Article : Google Scholar : PubMed/NCBI | |
|
Mukherjee A, Chiang CY, Daifotis HA, Nieman KM, Fahrmann JF, Lastra RR, Romero IL, Fiehn O and Lengyel E: Adipocyte-induced FABP4 expression in ovarian cancer cells promotes metastasis and mediates carboplatin resistance. Cancer Res. 80:1748–1761. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Liu X, Zhang P, Xu J, Lv G and Li Y: Lipid metabolism in tumor microenvironment: Novel therapeutic targets. Cancer Cell Int. 22:2242022. View Article : Google Scholar : PubMed/NCBI | |
|
Vasseur S and Guillaumond F: Lipids in cancer: A global view of the contribution of lipid pathways to metastatic formation and treatment resistance. Oncogenesis. 11:462022. View Article : Google Scholar : PubMed/NCBI | |
|
Mukherjee A, Bilecz AJ and Lengyel E: The adipocyte microenvironment and cancer. Cancer Metastasis Rev. 41:575–587. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Vlachostergios PJ: Loss of tumor suppressive properties of lipid metabolism enzyme CPT2 in ovarian carcinoma: Comment on 'CPT2 down-regulation promotes tumor growth and metastasis through inducing ROS/NFκB pathway in ovarian cancer' by Zhang et al. Transl Oncol. 14:1010672021. View Article : Google Scholar | |
|
Siegel RL, Miller KD and Jemal A: Cancer statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Gilks CB and Prat J: Ovarian carcinoma pathology and genetics: Recent advances. Hum Pathol. 40:1213–1223. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Montaigne D, Butruille L and Staels B: PPAR control of metabolism and cardiovascular functions. Nat Rev Cardiol. 18:809–823. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Luo X, Xu J, Yu J and Yi P: Shaping immune responses in the tumor microenvironment of ovarian cancer. Front Immunol. 12:6923602021. View Article : Google Scholar : PubMed/NCBI | |
|
Tian W, Lei N, Zhou J, Chen M, Guo R, Qin B, Li Y and Chang L: Extracellular vesicles in ovarian cancer chemoresistance, metastasis, and immune evasion. Cell Death Dis. 13:642022. View Article : Google Scholar : PubMed/NCBI | |
|
Li Y, Yu C and Deng W: Roles and mechanisms of adipokines in drug resistance of tumor cells. Eur J Pharmacol. 899:1740192021. View Article : Google Scholar : PubMed/NCBI | |
|
Dai L, Song K and Di W: Adipocytes: Active facilitators in epithelial ovarian cancer progression? J Ovarian Res. 13:1152020. View Article : Google Scholar : PubMed/NCBI | |
|
Chehade H, Tedja R, Ramos H, Bawa TS, Adzibolosu N, Gogoi R, Mor G and Alvero AB: Regulatory role of the adipose microenvironment on ovarian cancer progression. Cancers (Basel). 14:22672022. View Article : Google Scholar : PubMed/NCBI | |
|
Duan C, Yu M, Xu J, Li BY, Zhao Y and Kankala RK: Overcoming cancer multi-drug resistance (MDR): Reasons, mechanisms, nanotherapeutic solutions, and challenges. Biomed Pharmacother. 162:1146432023. View Article : Google Scholar : PubMed/NCBI | |
|
Pote MS and Gacche RN: ATP-binding cassette efflux transporters and MDR in cancer. Drug Discov Today. 28:1035372023. View Article : Google Scholar : PubMed/NCBI | |
|
Dean M, Moitra K and Allikmets R: The human ATP-binding cassette (ABC) transporter superfamily. Hum Mutat. 43:1162–1182. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Kukal S, Guin D, Rawat C, Bora S, Mishra MK, Sharma P, Paul PR, Kanojia N, Grewal GK, Kukreti S, et al: Multidrug efflux transporter ABCG2: Expression and regulation. Cell Mol Life Sci. 78:6887–6939. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Li B, Jiang J, Assaraf YG, Xiao H, Chen ZS and Huang C: Surmounting cancer drug resistance: New insights from the perspective of N6-methyladenosine RNA modification. Drug Resist Updat. 53:1007202020. View Article : Google Scholar | |
|
Mirza AZ, Althagafi II and Shamshad H: Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur J Med Chem. 166:502–513. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Ma S, Zhou B, Yang Q, Pan Y, Yang W, Freedland SJ, Ding LW, Freeman MR, Breunig JJ, Bhowmick NA, et al: A transcriptional regulatory loop of master regulator transcription factors, PPARG, and fatty acid synthesis promotes esophageal adenocarcinoma. Cancer Res. 81:1216–1229. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Mannan A, Garg N, Singh TG and Kang HK: Peroxisome proliferator-activated receptor-gamma (PPAR-γ): Molecular effects and its importance as a novel therapeutic target for cerebral ischemic injury. Neurochem Res. 46:2800–2831. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Szatmari I, Vámosi G, Brazda P, Balint BL, Benko S, Széles L, Jeney V, Ozvegy-Laczka C, Szántó A, Barta E, et al: Peroxisome proliferator-activated receptor gamma-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J Biol Chem. 281:23812–23823. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Lin Y, Bircsak KM, Gorczyca L, Wen X and Aleksunes LM: Regulation of the placental BCRP transporter by PPAR gamma. J Biochem Mol Toxicol. 31: View Article : Google Scholar : 2017. | |
|
Kim CE, Park HY, Won HJ, Kim M, Kwon B, Lee SJ, Kim DH, Shin JG and Seo SK: Repression of PPARγ reduces the ABCG2-mediated efflux activity of M2 macrophages. Int J Biochem Cell Biol. 130:1058952021. View Article : Google Scholar | |
|
Yu Z, Cai Y, Deng M, Li D, Wang X, Zheng H, Xu Y, Li W and Zhang W: Fat extract promotes angiogenesis in a murine model of limb ischemia: A novel cell-free therapeutic strategy. Stem Cell Res Ther. 9:2942018. View Article : Google Scholar : PubMed/NCBI | |
|
Yu C, Niu X, Du Y, Chen Y, Liu X, Xu L, Iwakura Y, Ma X, Li Y, Yao Z and Deng W: IL-17A promotes fatty acid uptake through the IL-17A/IL-17RA/p-STAT3/FABP4 axis to fuel ovarian cancer growth in an adipocyte-rich microenvironment. Cancer Immunol Immunother. 69:115–126. 2020. View Article : Google Scholar | |
|
Xu S, Yu C, Ma X, Li Y, Shen Y, Chen Y, Huang S, Zhang T, Deng W and Wang Y: IL-6 promotes nuclear translocation of HIF-1α to aggravate chemoresistance of ovarian cancer cells. Eur J Pharmacol. 894:1738172021. View Article : Google Scholar | |
|
Shen M, Xu Z, Xu W, Jiang K, Zhang F, Ding Q, Xu Z and Chen Y: Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J Exp Clin Cancer Res. 38:1492019. View Article : Google Scholar : PubMed/NCBI | |
|
Li D, Wang Y, Dong C, Chen T, Dong A, Ren J, Li W, Shu G, Yang J, Shen W, et al: CST1 inhibits ferroptosis and promotes gastric cancer metastasis by regulating GPX4 protein stability via OTUB1. Oncogene. 42:83–98. 2023. View Article : Google Scholar : | |
|
Christofides A, Konstantinidou E, Jani C and Boussiotis VA: The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism. 114:1543382021. View Article : Google Scholar | |
|
Hamilton TC, Young RC, McKoy WM, Grotzinger KR, Green JA, Chu EW, Whang-Peng J, Rogan AM, Green WR and Ozols RF: Characterization of a human ovarian carcinoma cell line (NIH:OVCAR-3) with androgen and estrogen receptors. Cancer Res. 43:5379–5389. 1983.PubMed/NCBI | |
|
Mitra AK, Davis DA, Tomar S, Roy L, Gurler H, Xie J, Lantvit DD, Cardenas H, Fang F, Liu Y, et al: In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecol Oncol. 138:372–377. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Zhao G, Tan Y, Cardenas H, Vayngart D, Wang Y, Huang H, Keathley R, Wei JJ, Ferreira CR, Orsulic S, et al: Ovarian cancer cell fate regulation by the dynamics between saturated and unsaturated fatty acids. Proc Natl Acad Sci USA. 119:e22034801192022. View Article : Google Scholar : PubMed/NCBI | |
|
Šrámek J, Němcová-Fürstová V and Kovář J: Molecular mechanisms of apoptosis induction and its regulation by fatty acids in pancreatic β-cells. Int J Mol Sci. 22:42852021. View Article : Google Scholar | |
|
Krümmel B, von Hanstein AS, Plötz T, Lenzen S and Mehmeti I: Differential effects of saturated and unsaturated free fatty acids on ferroptosis in rat β-cells. J Nutr Biochem. 106:1090132022. View Article : Google Scholar | |
|
Hoy AJ, Nagarajan SR and Butler LM: Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat Rev Cancer. 21:753–766. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Aggarwal S, Verma SS, Aggarwal S and Gupta SC: Drug repurposing for breast cancer therapy: Old weapon for new battle. Semin Cancer Biol. 68:8–20. 2021. View Article : Google Scholar | |
|
Chen HJ, Chung YL, Li CY, Chang YT, Wang CCN, Lee HY, Lin HY and Hung CC: Taxifolin Resensitizes multidrug resistance cancer cells via uncompetitive inhibition of P-glycoprotein function. Molecules. 23:30552018. View Article : Google Scholar : PubMed/NCBI | |
|
Engle K and Kumar G: Cancer multidrug-resistance reversal by ABCB1 inhibition: A recent update. Eur J Med Chem. 239:1145422022. View Article : Google Scholar : PubMed/NCBI | |
|
Modi A, Roy D, Sharma S, Vishnoi JR, Pareek P, Elhence P, Sharma P and Purohit P: ABC transporters in breast cancer: Their roles in multidrug resistance and beyond. J Drug Target. 30:927–947. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Wang J, Yang DH, Yang Y, Wang JQ, Cai CY, Lei ZN, Teng QX, Wu ZX, Zhao L and Chen ZS: Overexpression of ABCB1 transporter confers resistance to mTOR inhibitor WYE-354 in cancer cells. Int J Mol Sci. 21:13872020. View Article : Google Scholar : PubMed/NCBI | |
|
Bukowski K, Kciuk M and Kontek R: Mechanisms of multidrug resistance in cancer chemotherapy. Int J Mol Sci. 21:32332020. View Article : Google Scholar : PubMed/NCBI | |
|
He ZX, Zhao TQ, Gong YP, Zhang X, Ma LY and Liu HM: Pyrimidine: A promising scaffold for optimization to develop the inhibitors of ABC transporters. Eur J Med Chem. 200:1124582020. View Article : Google Scholar : PubMed/NCBI | |
|
Miyata H, Takada T, Toyoda Y, Matsuo H, Ichida K and Suzuki H: Identification of febuxostat as a new strong ABCG2 inhibitor: Potential applications and risks in clinical situations. Front Pharmacol. 7:5182016. View Article : Google Scholar | |
|
Deng F, Sjostedt N, Santo M, Neuvonen M, Niemi M and Kidron H: Novel inhibitors of breast cancer resistance protein (BCRP, ABCG2) among marketed drugs. Eur J Pharm Sci. 181:1063622023. View Article : Google Scholar | |
|
Cao Y: Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest. 129:3006–3017. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Zhou X, Zhang J, Lv W, Zhao C, Xia Y, Wu Y and Zhang Q: The pleiotropic roles of adipocyte secretome in remodeling breast cancer. J Exp Clin Cancer Res. 41:2032022. View Article : Google Scholar : PubMed/NCBI | |
|
Dumas JF and Brisson L: Interaction between adipose tissue and cancer cells: Role for cancer progression. Cancer Metastasis Rev. 40:31–46. 2021. View Article : Google Scholar | |
|
Brown KA and Scherer PE: Update on adipose tissue and cancer. Endocr Rev. 44:961–974. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Tewari S, Vargas R and Reizes O: The impact of obesity and adipokines on breast and gynecologic malignancies. Ann N Y Acad Sci. 1518:131–150. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Clemente-Suárez VJ, Redondo-Flórez L, Beltrán-Velasco AI, Martín-Rodríguez A, Martínez-Guardado I, Navarro-Jiménez E, Laborde-Cárdenas CC and Tornero-Aguilera JF: The role of adipokines in health and disease. Biomedicines. 11:12902023. View Article : Google Scholar : PubMed/NCBI | |
|
Rybinska I, Mangano N, Tagliabue E and Triulzi T: Cancer-associated adipocytes in breast cancer: Causes and consequences. Int J Mol Sci. 22:37752021. View Article : Google Scholar : PubMed/NCBI | |
|
Wróblewski M, Szewczyk-Golec K, Hołyńska-Iwan I, Wróblewska J and Woźniak A: Characteristics of selected adipokines in ascites and blood of ovarian cancer patients. Cancers (Basel). 13:47022021. View Article : Google Scholar : PubMed/NCBI | |
|
Rajesh Y and Sarkar D: Association of adipose tissue and adipokines with development of obesity-induced liver cancer. Int J Mol Sci. 22:21632021. View Article : Google Scholar : PubMed/NCBI | |
|
Conze D, Weiss L, Regen PS, Bhushan A, Weaver D, Johnson P and Rincon M: Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 61:8851–8858. 2001.PubMed/NCBI | |
|
Lipsey CC, Harbuzariu A, Robey RW, Huff LM, Gottesman MM and Gonzalez-Perez RR: Leptin signaling affects survival and chemoresistance of estrogen receptor negative breast cancer. Int J Mol Sci. 21:37942020. View Article : Google Scholar : PubMed/NCBI | |
|
Weng C, Dong H, Bai R, Sheng J, Chen G, Ding K, Lin W, Chen J and Xu Z: Angiogenin promotes angiogenesis via the endonucleolytic decay of miR-141 in colorectal cancer. Mol Ther Nucleic Acids. 27:1010–1022. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Lehuédé C, Li X, Dauvillier S, Vaysse C, Franchet C, Clement E, Esteve D, Longué M, Chaltiel L, Le Gonidec S, et al: Adipocytes promote breast cancer resistance to chemotherapy, a process amplified by obesity: Role of the major vault protein (MVP). Breast Cancer Res. 21:72019. View Article : Google Scholar | |
|
Sarkanen JR, Kaila V, Mannerström B, Räty S, Kuokkanen H, Miettinen S and Ylikomi T: Human adipose tissue extract induces angiogenesis and adipogenesis in vitro. Tissue Eng Part A. 18:17–25. 2012. View Article : Google Scholar | |
|
Amor S, Iglesias-de la Cruz MC, Ferrero E, Garcia-Villar O, Barrios V, Fernandez N, Monge L, García-Villalón AL and Granado M: Peritumoral adipose tissue as a source of inflammatory and angiogenic factors in colorectal cancer. Int J Colorectal Dis. 31:365–375. 2016. View Article : Google Scholar | |
|
Bejarano L, Jordāo MJC and Joyce JA: Therapeutic targeting of the tumor microenvironment. Cancer Discov. 11:933–959. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Huang M, Lin Y, Wang C, Deng L, Chen M, Assaraf YG, Chen ZS, Ye W and Zhang D: New insights into antiangiogenic therapy resistance in cancer: Mechanisms and therapeutic aspects. Drug Resist Updat. 64:1008492022. View Article : Google Scholar : PubMed/NCBI | |
|
Qi S, Deng S, Lian Z and Yu K: Novel Drugs with high efficacy against tumor angiogenesis. Int J Mol Sci. 23:69342022. View Article : Google Scholar : PubMed/NCBI | |
|
Iwamoto H, Abe M, Yang Y, Cui D, Seki T, Nakamura M, Hosaka K, Lim S, Wu J, He X, et al: Cancer lipid metabolism confers antiangiogenic drug resistance. Cell Metab. 28:104–117.e5. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Mentoor I, Engelbrecht AM, van Jaarsveld PJ and Nell T: Chemoresistance: Intricate interplay between breast tumor cells and adipocytes in the tumor microenvironment. Front Endocrinol (Lausanne). 9:7582018. View Article : Google Scholar | |
|
Bougaret L, Delort L, Billard H, Le Huede C, Boby C, De la Foye A, Rossary A, Mojallal A, Damour O, Auxenfans C, et al: Adipocyte/breast cancer cell crosstalk in obesity interferes with the anti-proliferative efficacy of tamoxifen. PLoS One. 13:e01915712018. View Article : Google Scholar : PubMed/NCBI | |
|
Yang J, Zaman MM, Vlasakov I, Roy R, Huang L, Martin CR, Freedman SD, Serhan CN and Moses MA: Adipocytes promote ovarian cancer chemoresistance. Sci Rep. 9:133162019. View Article : Google Scholar : PubMed/NCBI | |
|
Kasonga A, Kruger MC and Coetzee M: Activation of PPARs modulates signalling pathways and expression of regulatory genes in osteoclasts derived from human CD14+ monocytes. Int J Mol Sci. 20:17982019. View Article : Google Scholar : PubMed/NCBI | |
|
Li G, Li X, Mahmud I, Ysaguirre J, Fekry B, Wang S, Wei B, Eckel-Mahan KL, Lorenzi PL, Lehner R and Sun K: Interfering with lipid metabolism through targeting CES1 sensitizes hepatocellular carcinoma for chemotherapy. JCI Insight. 8:e1636242023. View Article : Google Scholar : | |
|
Schlotterbeck J, Cebo M, Kolb A and Lämmerhofer M: Quantitative analysis of chemoresistance-inducing fatty acid in food supplements using UHPLC-ESI-MS/MS. Anal Bioanal Chem. 411:479–491. 2019. View Article : Google Scholar | |
|
Zou Y, Watters A, Cheng N, Perry CE, Xu K, Alicea GM, Parris JLD, Baraban E, Ray P, Nayak A, et al: Polyunsaturated fatty acids from astrocytes activate PPARγ signaling in cancer cells to promote brain metastasis. Cancer Discov. 9:1720–1735. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Liotti A, Cosimato V, Mirra P, Cali G, Conza D, Secondo A, Luongo G, Terracciano D, Formisano P, Beguinot F, et al: Oleic acid promotes prostate cancer malignant phenotype via the G protein-coupled receptor FFA1/GPR40. J Cell Physiol. 233:7367–7378. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, Kim IS, Haldeman P, Mondal C, Yong-Gonzales V, et al: Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. 8:1006–1025. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang Y, Wang D, Lv B, Hou X, Liu Q, Liao C, Xu R, Zhang Y, Xu F and Zhang P: Oleic acid and insulin as key characteristics of T2D promote colorectal cancer deterioration in xenograft mice revealed by functional metabolomics. Front Oncol. 11:6850592021. View Article : Google Scholar : PubMed/NCBI | |
|
Li X, Ycaza J and Blumberg B: The environmental obesogen tributyltin chloride acts via peroxisome proliferator activated receptor gamma to induce adipogenesis in murine 3T3-L1 preadipocytes. J Steroid Biochem Mol Biol. 127:9–15. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Hernandez-Quiles M, Broekema MF and Kalkhoven E: PPARgamma in metabolism, immunity, and cancer: Unified and diverse mechanisms of action. Front Endocrinol (Lausanne). 12:6241122021. View Article : Google Scholar : PubMed/NCBI | |
|
Mal S, Dwivedi AR and Kumar V, Kumar N, Kumar B and Kumar V: Role of peroxisome proliferator-activated receptor gamma (PPARγ) in different disease states: Recent updates. Curr Med Chem. 28:3193–3215. 2021. View Article : Google Scholar | |
|
Zhang GY, Ahmed N, Riley C, Oliva K, Barker G, Quinn MA and Rice GE: Enhanced expression of peroxisome proliferator-activated receptor gamma in epithelial ovarian carcinoma. Br J Cancer. 92:113–119. 2005. View Article : Google Scholar | |
|
Al-Alem L, Southard RC, Kilgore MW and Curry TE: Specific thiazolidinediones inhibit ovarian cancer cell line proliferation and cause cell cycle arrest in a PPARγ independent manner. PLoS One. 6:e161792011. View Article : Google Scholar | |
|
Shin SJ, Kim JY, Kwon SY, Mun KC, Cho CH and Ha E: Ciglitazone enhances ovarian cancer cell death via inhibition of glucose transporter-1. Eur J Pharmacol. 743:17–23. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang Y, Ba Y, Liu C, Sun G, Ding L, Gao S, Hao J, Yu Z, Zhang J, Zen K, et al: PGC-1alpha induces apoptosis in human epithelial ovarian cancer cells through a PPARgamma-dependent pathway. Cell Res. 17:363–373. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Kim S, Lee JJ and Heo DS: PPARγ ligands induce growth inhibition and apoptosis through p63 and p73 in human ovarian cancer cells. Biochem Biophys Res Commun. 406:389–395. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Yokoyama Y, Xin B, Shigeto T and Mizunuma H: Combination of ciglitazone, a peroxisome proliferator-activated receptor gamma ligand, and cisplatin enhances the inhibition of growth of human ovarian cancers. J Cancer Res Clin Oncol. 137:1219–1228. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Cheng S, Qian K, Wang Y, Wang G, Liu X, Xiao Y and Wang X: PPARγ inhibition regulates the cell cycle, proliferation and motility of bladder cancer cells. J Cell Mol Med. 23:3724–3736. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Wu B, Sun X, Gupta HB, Yuan B, Li J, Ge F, Chiang HC, Zhang X, Zhang C, Zhang D, et al: Adipose PD-L1 modulates PD-1/PD-L1 checkpoint blockade immunotherapy efficacy in breast cancer. Oncoimmunology. 7:e15001072018. View Article : Google Scholar : PubMed/NCBI |