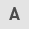Targeting mitochondrial bioenergetics by combination treatment with imatinib and dichloroacetate in human erythroleukemic K‑562 and colorectal HCT‑116 cancer cells
- Authors:
- Published online on: February 27, 2024 https://doi.org/10.3892/ijo.2024.5630
- Article Number: 42
-
Copyright: © Kakafika et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Cancer (tumor malignant) cells can appear and expand in several tissues as a result of genetic, epigenetic and developmental aberrations of the physiological homeostatic cell regulation (1). Such events can lead to a malignancy that is characterized by lack of cell differentiation and apoptosis, unlimited proliferation autonomy and self-renewal, insensitivity to external regulatory stimuli, angiogenesis and metastasis (2). Moreover, cancer cells undergo severe mitochondrial bioenergetic dysregulation (3-6) (Fig. 1). This dysfunction affects ATP production and increases the formation of reactive oxygen species (ROS) that promote mitochondrial oxidative stress and DNA damage (7) in leukemia (8) and other types of cancer.
ROS are mainly generated by complex I via the NADH:NAD ratio in mitochondria (9). While normal cells produce energy through the two major metabolic pathways [oxidative phosphorylation (OxPhos) and glycolysis], several cancer cells exhibit the well-discussed 'Warburg effect' (10,11), that characterizes chimeric energy generation states that oscillate between OxPhos and aerobic glycolysis, while accompanied by hypoxic-redox signaling (Fig. 1). In fact, while normal cells can generate more than 30 molecules of ATP per molecule of glucose via OxPhos, their cancer counterparts can produce 4 molecules of ATP, resulting in a net gain of 2 molecules of ATP per molecule of glucose consumed. In the same context, cancer cells tend to produce lactate to a markedly greater extent as compared with normal cells (12). This metabolic shift in mitochondria, known otherwise also as 'metabolic reprogramming', primes hypoxia and is likely related to alterations in the aberrant TP53 gene that is found mutated in ~50% of the malignant tumors (13).
As TP53 encodes a master transcription factor that regulates, among others, the expression of the mitochondrial protein for synthesis of cytochrome c oxidase 2 (SCO2) which is involved in the transfer of copper into the cytochrome c oxidase (COX) II subunit (14), mutations of TP53 appear to directly influence the dynamics of SCO2 in the holoenzyme complex IV biosynthesis and the assembly of COX in the mitochondrial respiratory chain (15). Encoded mutations in TP53 frequently drive mitochondrial abnormalities in cancer cells (16), while certain SCO2 mutations can lead to severe COX deficiency and contribute to various grades of mitochondrial dysfunction (15,17,18).
In a previous study conducted by the authors concerning human K-562 leukemia-based studies in 2014, it was noticed that imatinib (IM) mesylate (Gleevec), a potent inhibitor of the hybrid Bcr-Abl tyrosine phosphokinase activity, that is used as an effective antineoplastic drug for the treatment of human chronic myelogenous leukemia (CML) (19-23), downregulated the expression of the SCO2 and frataxin (FXN) genes (24). The latter encodes two important proteins involved in the heme-dependent cytochrome c oxidase biosynthesis and assembly pathway (HDCBAP) of COX in the mitochondria. This finding, along with the facts that IM has been found to promote apoptosis via a ROS-dependent JNK-Bcl2 mediated signaling pathway in melanoma cells (25) and that ROS activates the apoptosis signaling (26), suggested that IM probably affects the mitochondrial oxidative metabolism by ROS as well (27).
Cancer cells exhibit hypoxic profiles through metabolic changes (28) and produce energy preferentially via aerobic glycolysis (4). On those grounds, it was investigated whether co-treatment of cultured cancer cells with IM and dichloroacetate (DCA), could induce an increased anti-proliferative effect in the human K-562 leukemia and HCT-116 colorectal cell lines. DCA is a potent blocker of glycolysis through the inhibition of the pyruvate dehydrogenase (PDH) kinase (PDK) (29-31). DCA inhibits PDK and thus releases PDH, the rate-limiting enzyme for glucose oxidation. PDH converts the glycolysis-produced pyruvate that enters mitochondria to acetyl coenzyme A (acetyl-CoA) that is consumed in the processes of tricarboxylic acid cycle (TCA, also known as Krebs cycle) and OxPhos, thereby increasing the influx of acetyl-CoA from glycolysis into the TCA cycle. These two cell lines (K-562 and HCT-116) represent model systems for CML and a solid tumor (colorectal carcinoma), respectively, to examine the impact of: i) Chemoresistance of erythroleukemia K-562 cells to IM, as well as ii) the absence of full length p53 protein on the efficiency of the IM and DCA combination chemotherapy in colorectal carcinoma cells.
Thus, cultured cells of each cell line were independently treated either with IM or DCA alone, or in combination, and cell viability, inhibition of cell proliferation and the levels of several other process biomarkers (Table I) which include: i) the SCO2 and FXN proteins as markers of OxPhos (24,32), ii) the glycolytic mediators glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (33), pyruvate kinase M2 (PKM2) (34) as well as lactate dehydrogenase A (LDHA), a key enzyme for the 'Warburg effect' and lactate production (35-38), iii) the hypoxia dependent factors hypoxia inducing factor-1a (HIF-1a) (39,40) and SCF (41,42) and iv) the Redox signaling mediators heme oxygenase-1 (HO-1) (43,44) and NF-κB (45-47) were assessed.
Table IA total of 10 proteins assessed by western blotting as biomarkers for oxidative phosphorylation, glycolysis and hypoxia/oxidative stress in the human erythroleukemic K-562 and colorectal HCT-116 cancer cells. A total of 10 candidates of interest explored as potential biomarkers of mitochondrial bioenergetics share physicochemical interactions by STRING analysis. Prior to exploring the expression profiles of the 10 species selected as potential mitochondrial bioenergetics biomarkers, STRING analysis was performed to investigate whether they share physicochemical interactions as revealed in Fig. S5. |
Moreover, the present study aimed to investigate the impact of each treatment on the levels of ROS production in K-562 cells. Protein expression profiling analysis was performed by western blotting (WB) for OxPhos, glycolysis and hypoxia/oxidative stress related protein markers and ROS accumulation. Overall, evidence that the targeting of mitochondrial dysfunction in bioenergetics with IM and DCA can offer an alternative novel therapeutic approach to eradicate cancer cells was provided. The findings presented in the results Part I section were obtained from studies with the K-562 and K-562R CML cells and in Part II with the HCT-116 (+/+p53) and HCT-116 (−/−p53) colorectal carcinoma cells, respectively.
Materials and methods
Cell culture and materials
Human Bcr(−) Abl(+) CML erythroleukemia K-562 (IM)-sensitive (48) and IM-resistant K-562R [cat. no. CRL3344; American Type Culture Collection] cells were seeded at 2-4×105 cells/ml in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.). IM resistance appears to be related to oxidative stress (49). Therefore, in order to maintain kinase resistance to IM, the stock of cultured K-562R cells was maintained in culture medium supplemented with 0.89 μM IM [Imatinib mesylate (IM/Gleevec®)] was made available by the Novartis International AG) and 10% glutamine (GlutaMAX™; Gibco; Thermo Fisher Scientific, Inc.) (50). The human colon carcinoma HCT-116 (+/+p53) and HCT-116 (−/−p53) isogenic cell lines were kindly donated by Professor Bert Vogelstein, M.D. (Johns Hopkins University School of Medicine) (51). The latter carry both TP53 alleles disrupted. As described in the original publication, the p53 gene was disrupted in HCT-116 cells by homologous recombination. Two promoter-less targeting vectors, each containing a geneticinor hygromycin-resistance gene in place of genomic p53 sequences, were transfected sequentially in HCT-116 cells to disrupt the two p53 alleles. The targeting vectors used were constructed so that the first codons of the drug resistance markers replaced the first codon of the TP53 gene within exon 2.
Verification of the HCT-116 isogenic cell lines status by PCR genotyping and WB
Prior to any assessments with the two HCT-116 cohorts, the TP53 gene status of the two HCT-116 cohorts was validated by preparing total RNA from both populations for cDNA PCR genotyping. Samples A and B were amplified from total RNA, 2.5 μg each, isolated from HCT-116 (+/+p53) and HCT-116 (−/−p53) cells, using TRIsure (Bioline), according to the manufacturer's protocol. cDNA was synthesized with the Superscript 1st strand system for RT-PCR (Invitrogen; Thermo Fisher Scientific, Inc. Life Technologies Inc.) and DreamTaq DNA Polymerase (Thermo Fisher Scientific, Inc.) was employed for the PCR reaction, using two different pairs of oligos (Fig. S1A). The first set included the 31-nucleotide forward primer (FP53B) annealing in exon 2 at position 15,957-15,987 bp that begins with the first codon of the TP53 gene ATG: 5′-ATG GAG GAG CCG CAG TCA GAT CCT AGC GTC G-3′ and the 36-nucleotide reverse primer (BP53B), 5′-TCA GTC TGA GTC AGG CCC TTC TGT CTT GAA CAT GAG-3′, annealing in exon 11 at position 22,907-22,942 bp (5′-CTC ATG TTC AAG ACA GAA GGG CCT GAC TCA GAC TGA-3′). The expected PCR product size for this set was 1,182 bp. The second set included the 20-nucleotide forward primer (p53-F1) annealing in exon 5 at position 17,593-17,612 bp: 5′-TCA GCA TCT TAT CCG AGT GG-3′ and the same reverse primer (BP53B) as aforementioned. The expected PCR product size for this second set was 610 bp. The PCR reaction conditions were as follows: 95°C for 2 min, 39 cycles of 95°C for 1 min, 61°C for 1 min and 72°C for 90 sec, followed by incubation at 72°C for 10 min. PCR products were analyzed via 0.5% agarose gel electrophoresis in 1X TAE solution and ethidium bromide staining, using 100 bp DNA molecular weight markers (NIPPON Genetics Europe GmbH). As illustrated in Fig. S1B, the HCT-116 (+/+p53) cells expressed, both a longer 1,182 bp (143-1,324) and a shorter 610 bp (715-1,324), TP53 transcript. By contrast, the HCT-116 (−/−p53) cells expressed only the shorter 610 bp (715-1,324) TP53 transcript. The expression of a truncated and shorter p53 by the HCT-116 (−/−p53) cells was also validated at the protein level through WB in samples of total cytoplasmic extract, derived from HCT-116 (+/+p53) and HCT-116 (−/−p53) cells (Fig. S1C). Due to the insertion of the transgenes within the first codon of exon 2 of the TP53 gene, the HCT-116 (−/−p53) cells cannot encode a full length p53 transcription factor with functional regulatory properties as the wild-type analogue (52). The proliferation kinetic capacity of the HCT-116 (+/+ p53) and HCT-116 (−/−p53) cells was additionally evaluated by seeding them at 1.5×105 cells/ml (Fig. S2). HCT-116 (+/+p53) and/or HCT-116 (−/−p53) cells were cultured in McCoy's medium (Gibco; Thermo Fisher Scientific, Inc.) and were harvested at 3×105 cells/ml for isolating the cytoplasmic fraction and at a 3×106 cells/ml density to isolate the mitochondrial fraction.
Determination of cell proliferation, cell viability and IC50 value
All cell cultures employed in the present study, were incubated in media supplemented with 10% v/v FBS (Gibco; Thermo Fisher Scientific, Inc.) and 1% v/v PS (penicillin-streptomycin with amphotericin; Gibco; Thermo Fisher Scientific, Inc.) at 37°C with 5% CO2 humidified atmosphere (~95%). IM (Gleevec) was kindly donated by the Novartis Pharma AG Oncology Group (Dr Paul Manley) to Professor Asterios S. Tsiftsoglou. IM was dissolved in distilled water and added in culture media at a concentration of 1-2 μM (just lower than the Cmax of 2.7 μM achieved in CML patients treated with 400 mg/day) (23). DCA was purchased from Sigma-Aldrich; Merck KGaA (cat. no. D54702). The stock solution was diluted in distilled water and added in cultures at a final concentration of 4 mM. Cell proliferation of untreated (control), IM-treated, DCA-treated and IM/DCA-treated cultures was determined at various time intervals by measuring their cell concentrations using a Neubauer hemocytometer under a light microscope. Cell viability was similarly assessed by using a trypan blue solution (0.4% w/v; Sigma-Aldrich; Merck KGaA) exclusion assay approach (53). This exclusion test gives a clear-cut kinetic profile on the percentage (%) of dead and alive cells in a given culture at any given moment. This information is critical to follow the kinetic analysis of the accumulation of dead cells in each culture exposed to any agent. The doubling time was estimated using the doubling time cell calculator (http://www.doubling-time.com/compute_more.php). In order to determine the corresponding IC50 value for each compound, each cell line was treated separately with increasing concentrations of IM or DCA for 24, 48 and 72 h. Detailed graphic illustrations of the IC50 studies are included in Figs. S3 and S4.
Isolation of cytoplasmic protein lysates
Cell culture suspensions were used for total protein isolation. Cells were incubated separately with either 1 or 2 μM IM, 4 mM DCA or in combination with these two agents for 72 h. Cell suspensions were harvested at a 60-80% confluency, washed twice with cold PBS (1X), centrifuged at 1,100 × g and the resulting pellets were lysed with RIPA buffer (1 M sodium phosphate, 5 M NaCl, 10% SDS, 10% NP-40 and 1% deoxycholic acid sodium salt, pH 7.2), supplemented with protease inhibitors cocktail (1X) (Sigma-Aldrich; Merck KGaA). After centrifugation at 10,000 × g for 15 min at 4°C, all supernatants were collected, aliquoted and stored at −20°C, as representative of the cellular total protein content. Untreated cell cultures incubated without any agent served as control experiments.
Subcellular fractionation and isolation of mitochondria protein lysates
The mitochondrial fraction was isolated using a mitochondria lysis buffer (10 mM Tris-HCl, 0.25 mM sucrose, 0.2 mM EDTA, pH 7.8), supplemented with protease inhibitors cocktail (1X) (Sigma-Aldrich; Merck KGaA). Lysed cells were centrifuged at 1,000 × g for 5 min at 4°C to pellet mitochondria and were deep frozen for 30 min at −70°C. This step was repeated three times. Defrosted samples were disrupted by repeatedly passages through a 21-gauge needle and centrifuged at 1,000 × g for 10 min at 4°C to pellet nuclei and cellular debris. Post-nuclear supernatant obtained was then centrifuged at 10,000 × g for 15 min at 4°C to collect intact mitochondria, which were resuspended in mitochondria storage buffer (Qproteome Mitochondria Isolation Kit; cat. no. 37612; Qiagen GmbH) for storage at −20°C and the analysis of mitochondrial proteins (SCO2 and FXN) by WB (54).
Western blot analysis
All protein lysates obtained were quantified for protein content using the Bradford reagent (Bio-Rad Laboratories, Inc.) (55). Samples of 40 to 80 μg (micrograms) from each protein fraction were used for further analysis. Protein samples were mixed with the appropriate volume of loading buffer (1X), heated at 95°C for 5 min, run on a 12-15% acrylamide SDS-PAGE gel and then transferred to PVDF membrane. To ensure that proteins were successfully transferred, the PVDF membrane was stained with Ponceau S Staining Solution (Sigma-Aldrich; Merck KGaA). All membranes were blocked with 5% Non-fat milk in PBST buffer (PBS with 0.1% Tween® 20) for 75 min at room temperature (RT) with gentle agitation and then washed thoroughly with PBST. The primary antibodies were diluted in 3% BSA with PBST buffer or 2% Non-fat milk with PBST buffer. All membranes were incubated with the primary antibodies for 75 min at RT or overnight at 4°C. The incubated membranes were then washed with 10 ml PBST for 15 min, followed by three more washes with 5 ml PBST for 5 min. The primary antibodies were detected by staining with secondary alkaline phosphatase (AP)-conjugated antibodies. The secondary antibodies were diluted in 2% non-fat milk with PBST buffer and staining was carried out for 75 min at RT followed by similar washes as earlier. Prior to AP detection, each membrane was washed with 10 ml of AP Buffer (100 mM Tris-HCl, 100 mM NaCl, 50 mM MgCl2, pH 9.5), followed by three additional washes with 5 ml for 5 min. For AP visualization, each membrane was transferred to 4 ml of AP Buffer containing BCIP (5-Bromo-4-Chloro-3-Indolyl Phosphatase) (cat. no. A1117; BioChemica; AppliChem GmbH) and NBT (Nitroblue Tetrazolium Chloride) (cat. no. 10008; Biotium, Inc.). Detection was carried out at RT until the distinct bands corresponding to the proteins of interest appeared. Immunoblotting was conducted using the primary antibodies listed in Table SI. In order to ascertain equal loading of the samples, membranes were also probed with a monoclonal anti-α-tubulin antibody or a monoclonal anti-β-actin antibody. Goat anti-rabbit IgG-AP (2:5,000; cat. no. AP132, Sigma-Aldrich; Merck KGaA) and goat anti-mouse IgG-AP (1:4,000; cat. no. sc-2008; Santa Cruz Biotechnology, Inc.) antibodies were used as secondary antibodies and the signal intensities on the membranes were assessed with BCIP/NBT staining. The protein molecular weight markers used MWP02 and MWP03 (NIPPON Genetics Europe GmbH) facilitated the molecular weight estimation. The ratio of the intensity of each protein band detected in relation to the corresponding intensity of either α-tubulin and/or β-actin proteins band for each sample examined was calculated using the Image Studio Lite 5.0 software (LI-COR Biosciences) and used for densitometry analysis of the blots. The data are presented as fold difference to control, using the GraphPad Prism 8.0.2 for Windows (GraphPad Software; Dotmatics) for illustration purposes.
Measurement of total ROS content in K-562 cells by flow cytometry
Intracellular ROS were measured in K-562 and K-562R cells by staining with the fluorogenic probe 2′,7′-dichlorofluorescein diacetate (DCFDA; cat no. D6883; Sigma-Aldrich; Merck KGaA) followed by flow cytometry analysis (56). Cultured cells were incubated at 37°C, 5% CO2 for 72 h, either with 1 or 2 μM IM, or 4 mM DCA, or a combination of IM/DCA. All incubated cells were harvested, diluted in PBS (1X) and subsequently treated with 20 μM_DCFDA for 20 min at 37°C without light. For ROS quantification the stained cells were collected by centrifugation at 150 × g for 5 min in RT and resuspended in 1 ml PBS. All cells were analyzed by flow cytometry detecting the increase in fluorescence at 550 nm after sample excitation at 485 nm on a CyFlow® Robby 8 Autoloading Station flow cytometer (Sysmex Partec GmbH) and the data was analyzed using the FlowJo_V10 software (BD Biosciences).
Network biology modelling
To uncover putative connections among the selected molecules of interest and biological pathways, the Enrichr Knowledge Graph web-based platform was used (https://enrichr-kg.dev.maayanlab.cloud/). A total of five biological pathways were leveraged from the Gene Ontology (https://geneontology.org/), Kyoto Encyclopedia of Genes and Genomes (https://www.genome.jp/kegg/), Wikipathways 2021 (https://www.wikipathways.org/), Reactome (https://reactome.org/) and DisGeNET (https://www.disgenet.org/) databases and a protein-pathway bipartite graph was retrieved. Visualization of this bipartite graph was accomplished through Cytoscape (https://cytoscape.org/). Furthermore, the STRING platform (https://string-db.org/) was used to uncover putative physicochemical protein-protein interactions among the molecules of interest (combined score >0.4) (57).
Statistical analysis
All experiments were conducted at least in triplicates (3x) and the statistical significance was assessed by using GraphPad Prism 8.0.2 for Windows (GraphPad Software; Dotmatics) or R programming language [version 3.2.2, R Core Team (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing (https://www.R-project.org/)]. All data are expressed as the mean ± SD and were analyzed by Kruskal-Wallis test, paired t-test (to assess if the differences between paired values are consistent) and repeated ANOVA measurements. P<0.05 was considered to indicate a statistically significant difference.
Results
Part I. Studies with K-562 and IM-resistant K-562R chronic myeloid leukemia cells
IM and DCA inhibit cell proliferation and promote cell death in both IM-sensitive K-562 and resistant K-562R cell lines
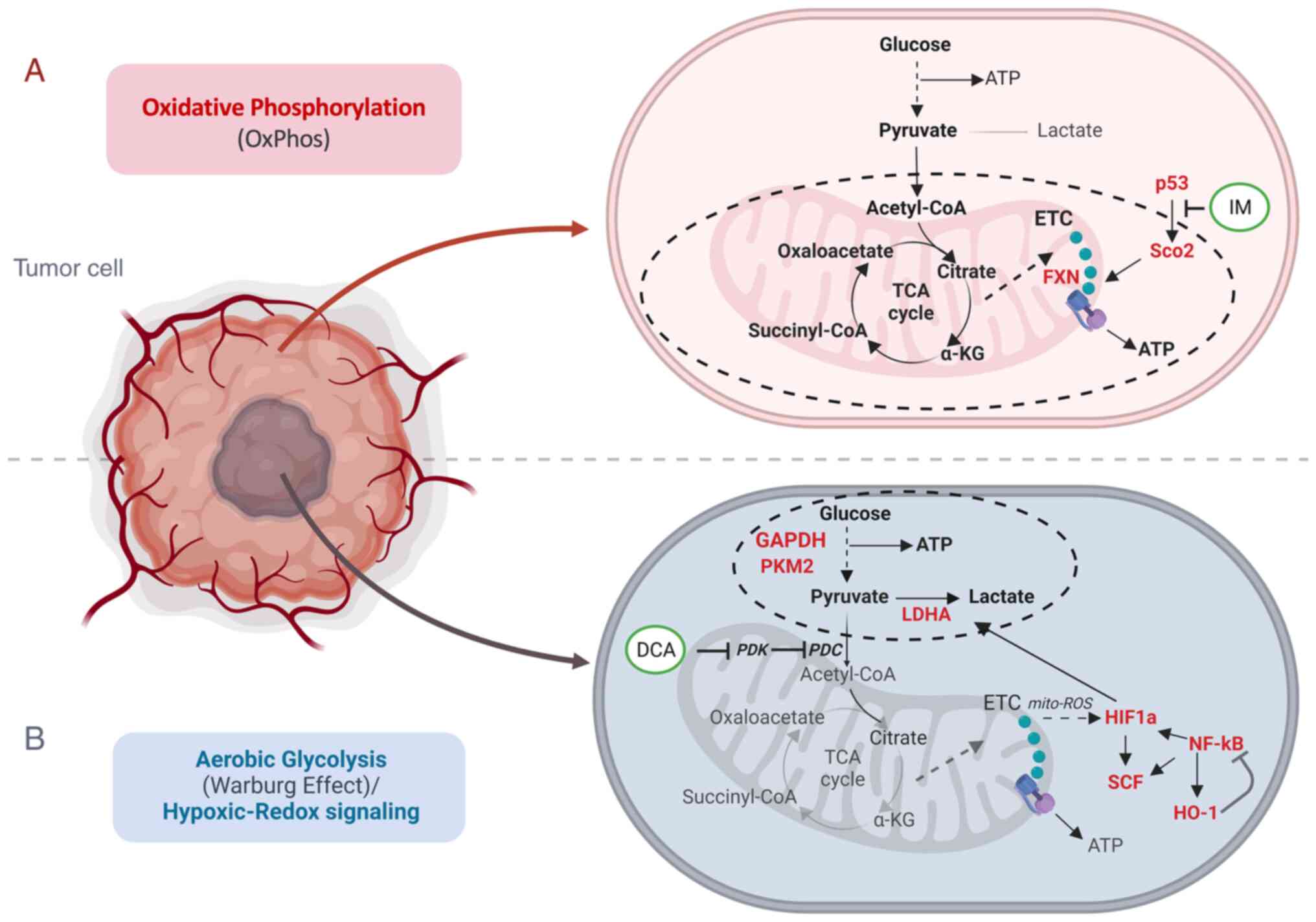
Preliminary experiments using K-562 and K-562R cultured cells were carried out to: i) Determine independently, after a 72-h incubation, the IC50 values for IM and DCA by applying increasing concentrations that ranged from μM to mM, respectively; ii) assess the proliferation behavior and kinetics of each cell line under treatment; and iii) determine the proportion of dead cells accumulated in each treated culture. These experiments revealed that the IC50 for IM was 0.3 μM for the K-562 cells and 6 μM (20-fold higher) for K-562R cells, while the IC50 value for DCA was calculated at 4 mM for both K-562 and K-562R cells (Fig. S3i-iv; Table SII). With respect to cell proliferation, treatment with 1-2 μM IM markedly reduced cell proliferation (~95%) in K-562 cells, but to a markedly less extent in IM-resistant K-562R cells, as expected (Fig. 2A and B). DCA added at 4 mM caused the same degree of proliferation inhibition in both cell types. Co-treatment of IM with DCA also markedly reduced cell proliferation by ~95% in the K-562 cells, but considerably less (~50%) in the K-562R cells (Fig. 2A and B). Regarding the ability of these agents to promote cell death, IM alone induced >60% cell death in K-562 cells (Fig. 2C), but <20% in K-562R cells (Fig. 2D). DCA, on the other hand, when applied at 4 mM generated elimination in ~40% of K-562 cells (Fig. 2C) and in <20% of IM-resistant K-562R cells (Fig. 2D). Co-exposure to IM/DCA induced cell death to >75% of K-562 cells (Fig. 2C), but to <30% of the K-562R cells (Fig. 2D). These data taken together indicated that treatment with both agents is considerably potent as it significantly reduced cell proliferation and induced cell death.
IM and DCA disturb OxPhos mediators SCO2 and FXN in opposing fashion depending on IM-resistance
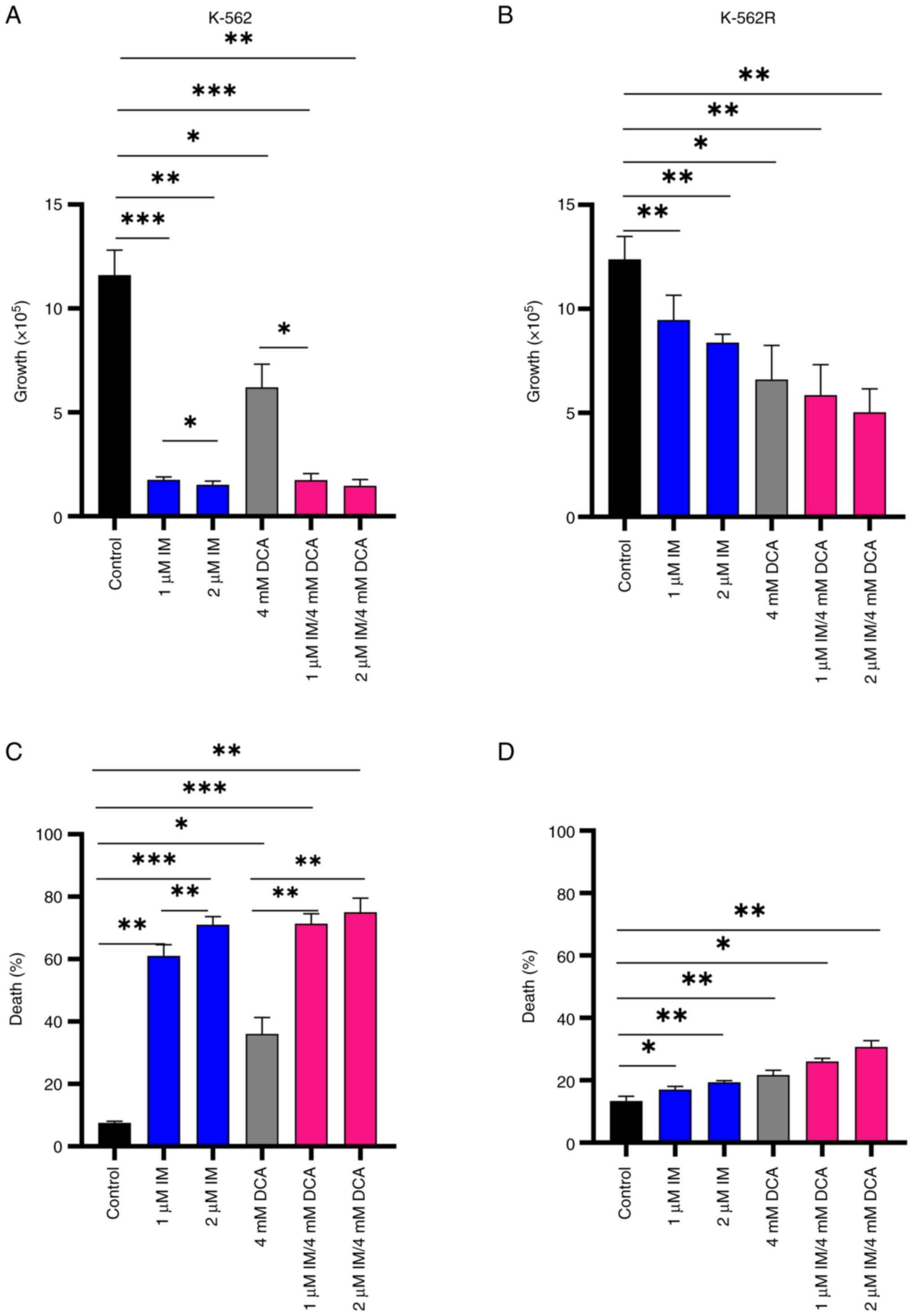
A previous study by the authors (24) in which IM downregulates the expression of SCO2 and FXN genes that represent biomarkers of OxPhos, prompted the authors to investigate whether, and to what extent, DCA alone or with IM can influence the expression of these two genes. The present study revealed that IM, either alone or with DCA, significantly suppressed the levels of both SCO2 and FXN proteins, by >85-90%, while DCA alone had no effect in suppressing the levels of both proteins in K-562 cells (Fig. 3A and B). In the K-562R cells, however, DCA was found to prime higher levels of SCO2 and FXN expression by up to 4-fold as compared with K-562 cells, presumably due to chemoresistance to IM. Finally, in the K-562R cells, the combination of IM with DCA (IM/DCA) led to considerable increases of SCO2 and FXN protein levels by 4 to 6-fold. In conclusion, these findings confirmed that when added together, IM/DCA, maintained the expression levels of OxPhos markers SCO2 and FXN in K-562 cells, but this effect was diminished in the K-562R cells as both were strongly upregulated.
IM and DCA affect the levels of the glycolytic biomarkers GAPDH, PKM2 and LDHA, but to a different extent in the K-562 and K-562R leukemia cells
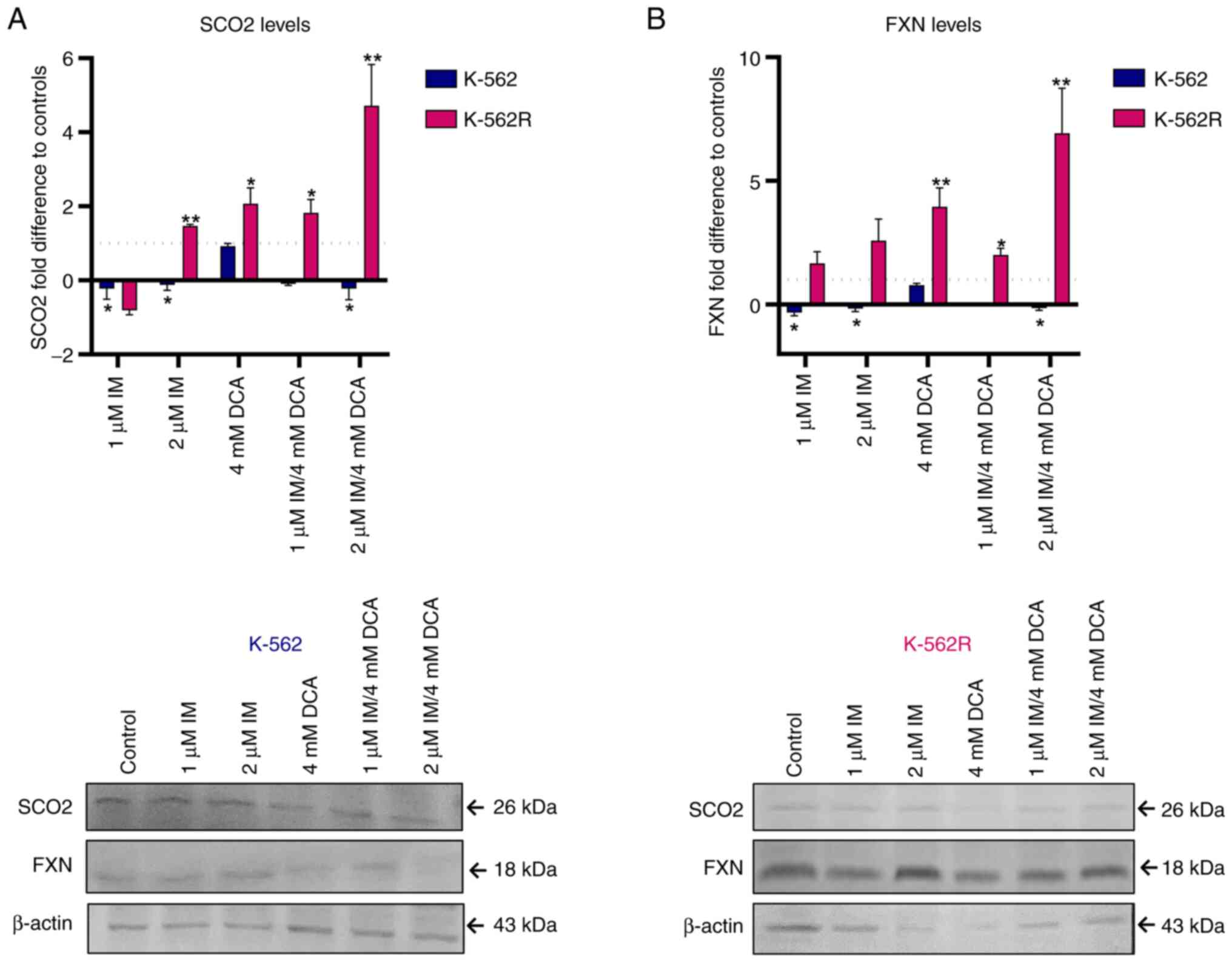
As demonstrated in Fig. 1, GAPDH, PKM2 and LDHA are three functionally important protein biomarkers that are highly involved in the glycolysis of cancer cells leading to the production of lactate. As revealed in Fig. 4A-C in the K-562 cells, IM alone or with DCA (IM/DCA) increased the protein levels of LDHA and GAPDH by 2 to 4-fold, with marginal effects on the levels of PKM2. In K-562R cells, however, when IM was added at 2 μM, alone or in combination with DCA, led to narrower increases of LDHA and GAPDH levels compared with K-562 cells. When IM was combined with DCA (IM/DCA), at 2 μM and 4 mM respectively, led to a moderate increase of PKM2 in K-562, but to a greater increase in the K-562R. The different degrees of inhibition of glycolytic markers observed between K-562 and K-562R may be attributed to chemoresistance factors.
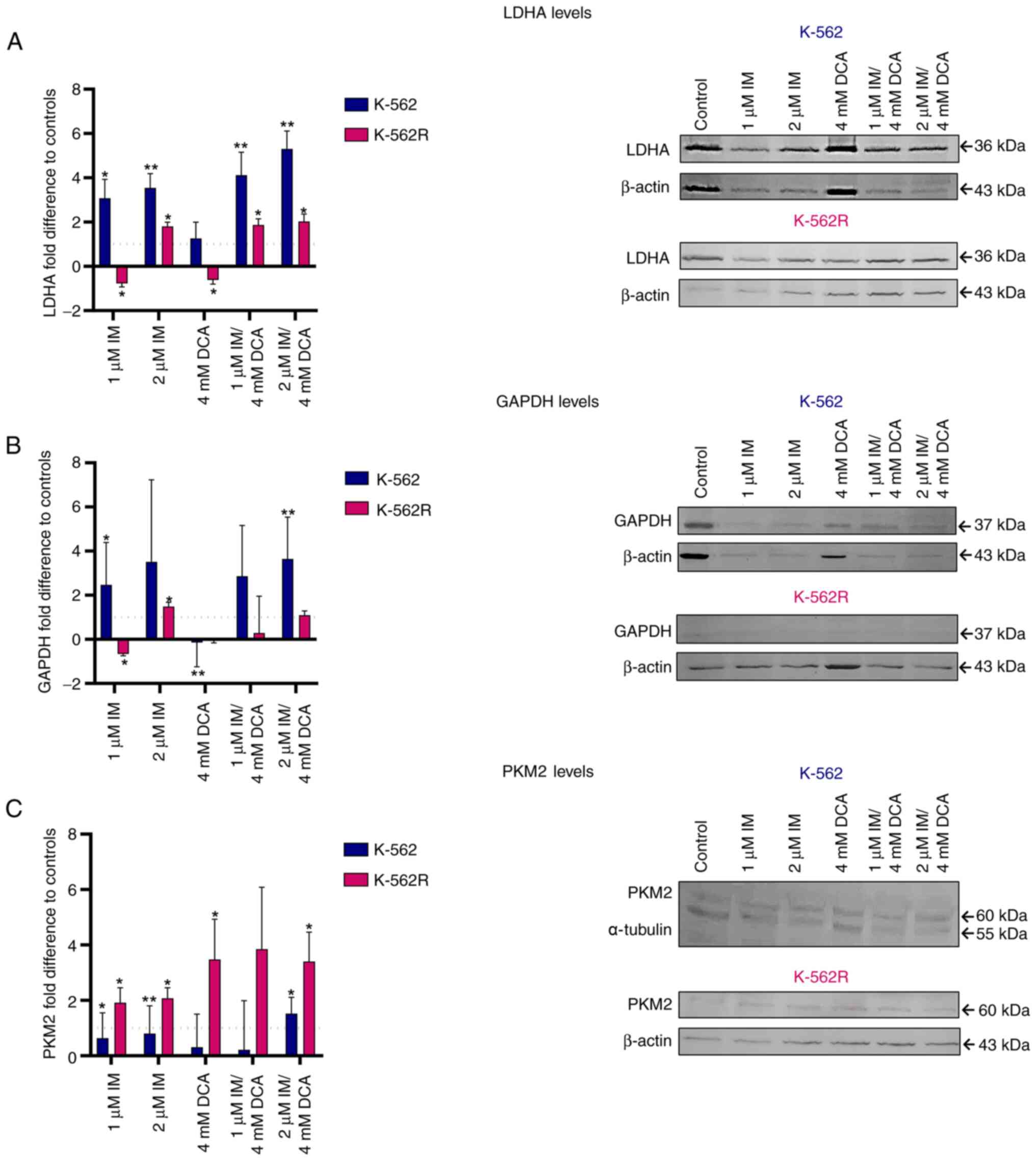
Effects of IM and DCA on the levels of hypoxic and oxidative stress response factors HIF-1a, NF-κB, SCF and vascular endothelial growth factor (VEGF)
As it is known that the majority of tumor cells are relatively hypoxic and undergo an oxidative stress response, the K-562 and K-562R IM/DCA treated cultures were assessed for the protein levels of transcription factors HIF-1a and NF-κB, as well as for SCF and VEGF. The results are revealed in Fig. 5A-D. In K-562 cells, IM increased HIF-1a protein levels by 5 to 10-fold. When both agents were used (IM/DCA), they increased the level of HIF-1a by ~5-fold (Fig. 5A) in K-562 cells. When K-562R were treated with IM alone or in combination with DCA (IM/DCA), similar, but smaller (~2-fold) increases of HIF-1a were observed. DCA alone increased HIF-1a by ~3-fold only in the K-562 cells. Concerning NF-κB, IM alone, as well as in combination with DCA, exerted a very mild decrease in K-562 protein levels (Fig. 5B). When the SCF protein levels were examined in K-562 cells, IM alone or in combination with DCA (IM/DCA), led to an increase of 2 to 4-fold (Fig. 5C). For the case of VEGF, IM and in combination with DCA (IM/DCA) increased its protein levels in the K-562 cells by 50 to 200-fold, while in the K-562R cells less than 2-fold. In conclusion, less extensive responses were recorded for the three factors (HIF-1a, SCF and VEGF) in the K-562R treated cells, as demonstrated in Fig. 5A, C and D, respectively.
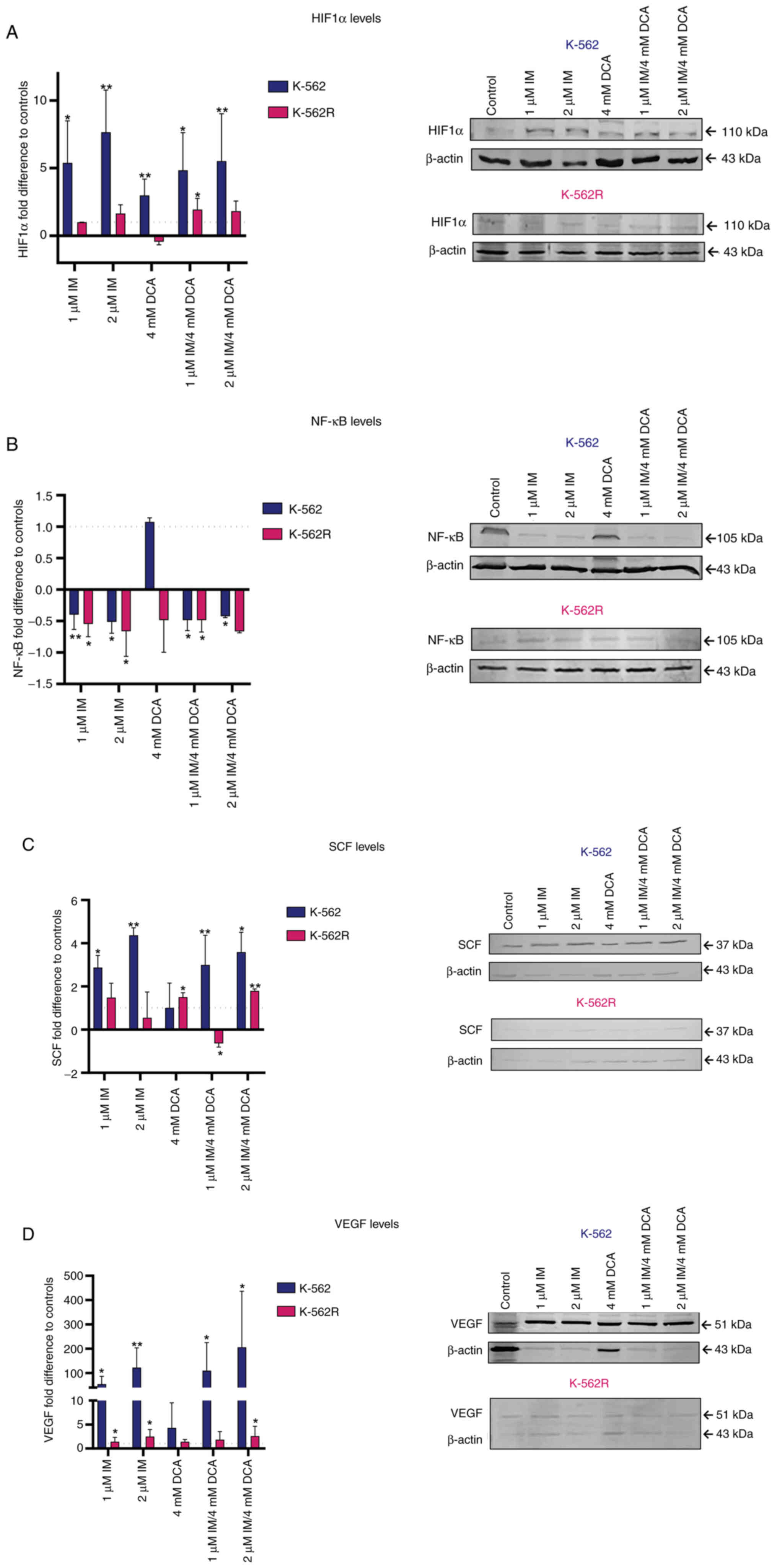
IM suppresses the levels of ROS in K-562 and K-562R cells, while DCA only in the non-resistant cells
In order to detect and subsequently measure the total content of ROS in K-562 and K-562R cells, it was explored whether these cells contain detectable ROS and subsequently it was examined whether IM, DCA or both agents affect their accumulation. Flow cytometric analysis using DCFDA as fluorescent probe was carried out. The results revealed that resting K-562 cells contained a relatively high level of ROS as revealed in Fig. 6A and that treatment with IM, or IM with DCA suppressed the levels of ROS to almost zero. In the K-562R cells however, which contained comparatively less ROS compared with the IM-sensitive K-562 cells, IM and the combination with DCA suppressed ROS to a small extent and not in the same levels with the K-562 cells (Figs. 6A and B; S6A and B). Notably, it was observed that treatment of the K-562R cells with DCA led to an increase of ROS, while treatment of the K-562 cells with DCA lowered, but did not flatten the levels of ROS (Figs. 6A and B; S6A and B). These data indicated that the K-562 and K-562R cell states are metabolically differentially tuned with higher levels of ROS in the K-562 cells. Challenging of glycolysis in the K-562R cells with DCA appears to promote an increase of ROS, probably as OxPhos is primed.
Part II. Studies with solid tumor HCT-116 (+/+p53) and HCT-116 (−/−p53) colorectal carcinoma cells
In the second part of the experiments, experiments regarding colorectal carcinoma HCT-116 (+/+p53) and HCT-116 (−/−p53) cells were conducted. Prior to starting the present comparative assessments, it was considered critical to confirm by PCR that the HCT-116 (−/−p53) cells, which contain the disrupted TP53 gene (51,58), are unable to produce the full length p53 transcription factor that regulates several genes, including SCO2. The presence of full length p53 protein in cells is critical for the production of SCO2 protein, necessary for the COX assembly and thus the overall functional performance of OxPhos. In addition, p53 and p21 proteins are also required to sustain growth arrest (51). After confirming that targeting of exon 2 of the TP53 gene leads to lack of a full length mature and functional p53 protein acting as a transcriptional activator for the SCO2 and FXN genes, as illustrated in Fig. S1, in vitro cell pharmacological studies with the two adherent HCT-116 colorectal carcinoma cell lines were carried out (Fig. S2). The doubling time of these cells were estimated to be 28 h for HCT-116 (+/+p53) cells, compared with 24 h for the HCT-116 (−/−p53) cells.
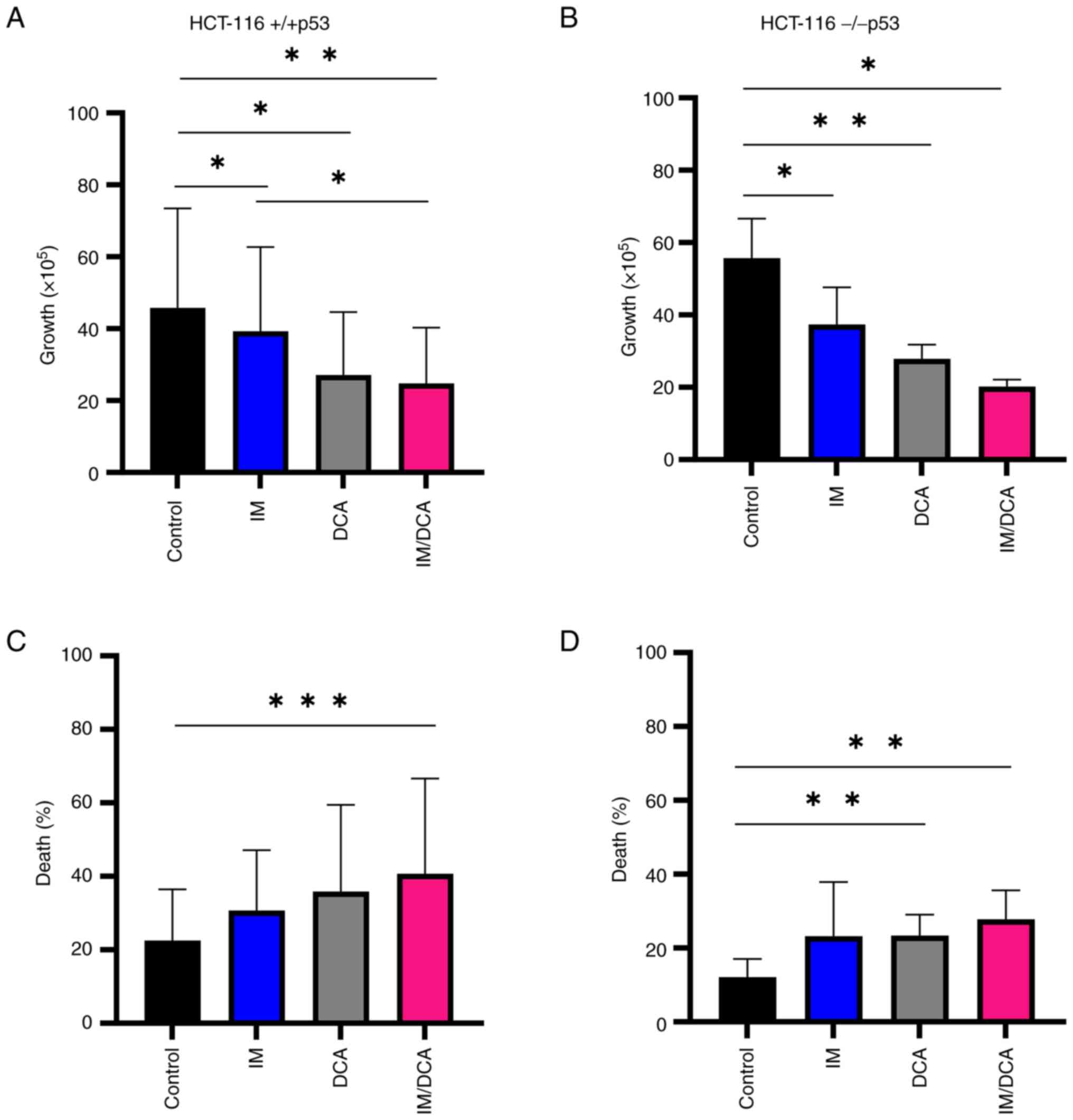
IM, DCA and their combination inhibit cell proliferation and promote cell death in the HCT-116 (+/+p53) and HCT-116 (−/−p53) cells
Preliminary experiments using HCT-116 (+/+p53) and HCT-116 (−/−p53) cultured cells were carried out to: i) Determine independently, after a 72-h incubation, the IC50 values for IM and DCA by applying increasing concentrations that ranged from μM to mM, respectively (Fig. S4i-iv; Table SII); ii) assess the proliferation behavior and kinetics of each cell line under treatment; and iii) determine the proportion of dead cells accumulated in each treated culture. The effects on cell proliferation and death by treating both HCT-116 cell lines with IM, DCA or IM/DCA are demonstrated in Fig. 7A-D. While IM or DCA caused moderate inhibition of cell proliferation, IM/DCA decreased cell proliferation by >50-60% in either cell line (Fig. 7A and B). Cell death in both HCT-116 cell lines increased after treatment with either agent alone, and nearly doubled by the combination of IM and DCA (Fig. 7C and D). This data indicated that all treatments inhibited cell proliferation, while cell death for both cell lines was significantly increased by the co-treatment with IM and DCA, regardless of any marginal differences observed between the two cell types (+/+p53) vs. (−/−p53).
IM, DCA and their combination affect the levels of the OxPhos SCO2 and FXN proteins in the HCT-116 (−/−p53) cells
After both HCT-116 cell lines were treated with IM, DCA or with a combinatorial treatment, the levels of SCO2 and FXN were assessed by gel electrophoresis and WB. In HCT-116 (+/+p53) cells, IM marginally decreased the levels of SCO2, while DCA as well as IM/DCA increased SCO2 by ~2-fold (Fig. 8A). As DCA is an inhibitor of glycolysis, an increase in the levels of SCO2 could be attributed to the shifting of the metabolism of HCT-116 cells, which relies primarily on glycolysis, towards OxPhos. In HCT-116 (−/−p53) cells, however, IM decreased the level of SCO2 mildly and similar behavior was observed by the IM/DCA co-treatment (Fig. 8A). Although DCA can inhibit glycolysis, due to the lack of the full length functional p53 protein (due to the absence of exon 2), the SCO2 protein levels did not change significantly. A similar analysis in the HCT-116 (+/+p53) cells demonstrated in Fig. 8B indicated that, similarly to SCO2, although IM decreased the FXN levels, DCA and IM/DCA co-treatment increased the level of frataxin considerably by almost ~4-fold for the case of the two agents. This may also be due to the fact that the FXN gene expression has been reported to be regulated by the transcription factor of TP53 (59). The same approach however, for the HCT-116 (−/−p53) cells, indicated that all IM and DCA treatments led to slight reduction of FXN protein levels (Fig. 8B). As aforementioned for the case of SCO2, this could be due to the lack of a full-length p53 protein in the HCT-116 (−/−p53) cells.
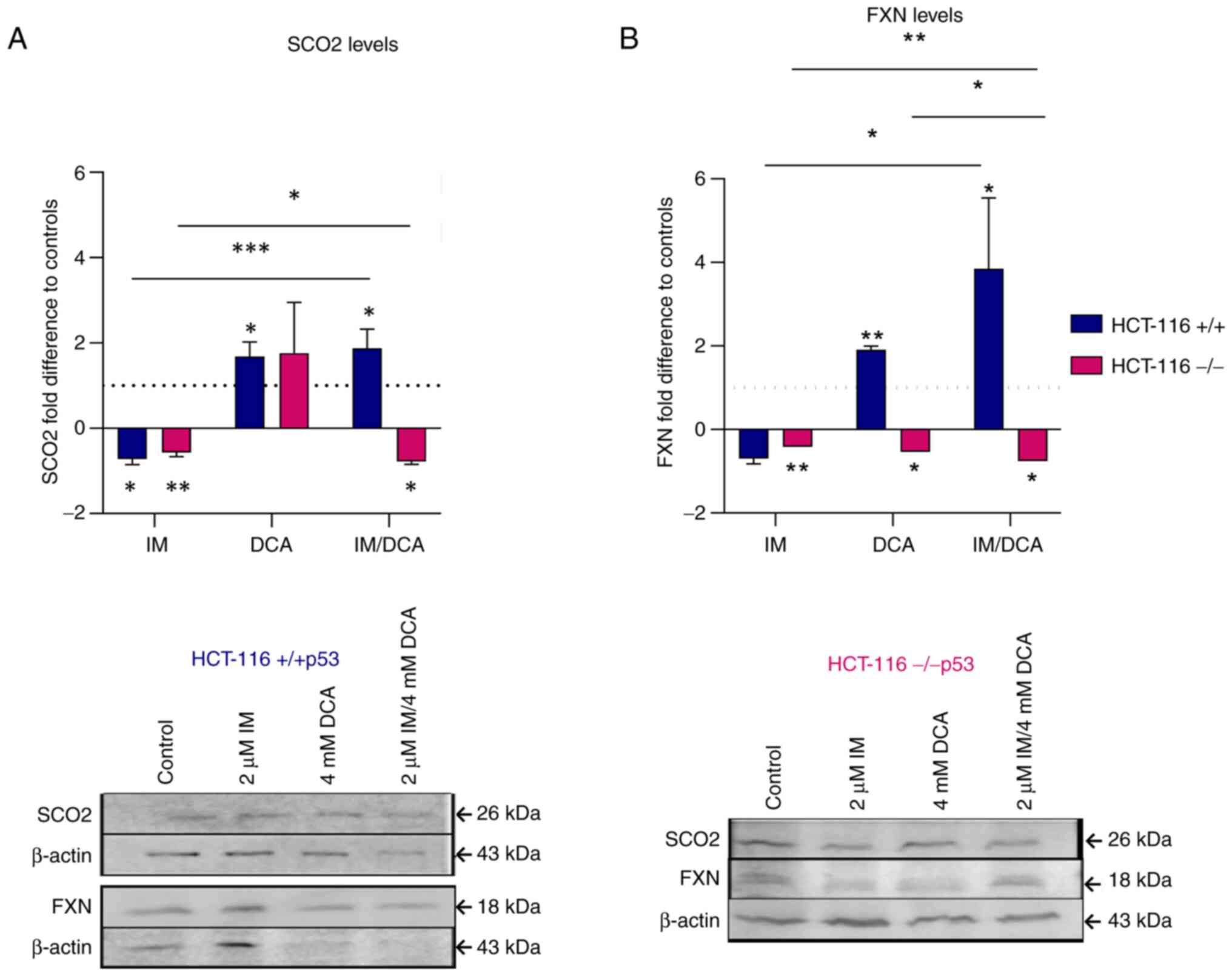
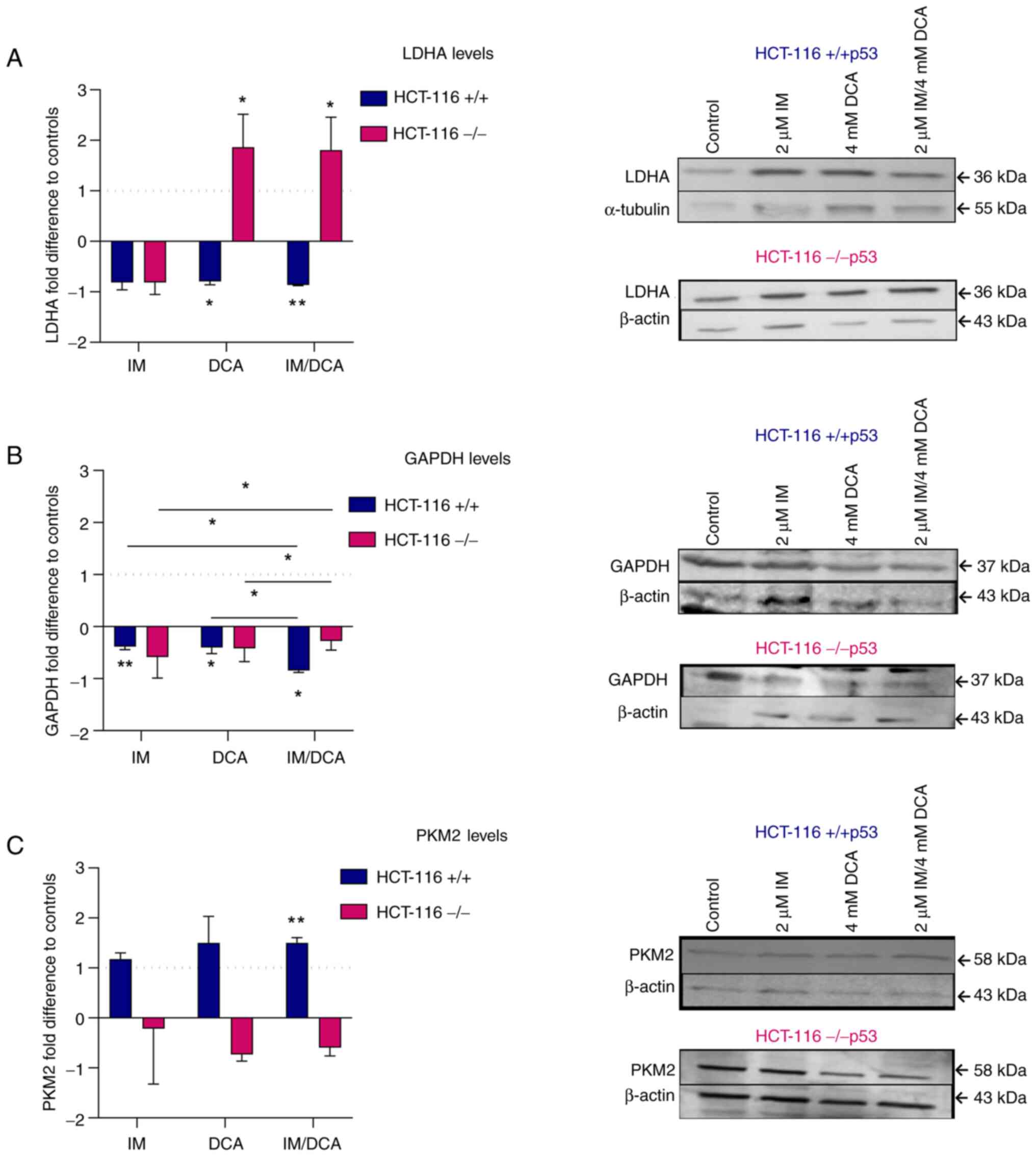
Treatment of HCT-116 cell lines with IM, DCA and their combination modulates the levels of glycolytic biomarkers
Concerning the protein levels of LDHA in IM-, DCA- or IM/DCA-treated HCT-116 (+/+p53) cells, an overall mild decrease was observed. Interestingly, the treatment of the HCT-116 (−/−p53) cells with DCA or IM/DCA increased the levels of LDHA by ~2-fold (Fig. 9A). The absence of full-length p53 protein appeared to play a crucial role in response of HCT-116 (−/−p53) cells to DCA. According to literature, p53 negatively regulates LDHA expression by directly binding in its promoter region (60). Furthermore, the p53/LDHA axis negatively regulates aerobic glycolysis and tumor progression in breast cancer expressing wild-type p53 (60). The treatment of all HCT-116 cells under study with IM, DCA or IM/DCA led to broad and mild reduction of the GAPDH protein levels (Fig. 9B). An increase of PKM2 was observed in the HCT-116 (+/+ p53) cells treated with IM, DCA or in the combinatorial treatment. By contrast, the same treatments induced very mild reductions of PKM2 in the HCT116 (−/−p53) cells (Fig. 9C). In conclusion, IM and DCA reduced the protein levels of LDHA and GAPDH in HCT-116 (+/+p53) cells, while in HCT-116 (−/−p53) cells DCA alone or in combination with IM (IM/DCA) reversed this for LDHA. The lack of a full length p53 in HCT-116 (−/−p53) cells cannot facilitate the increase of PKM2 upon stimulation with IM and DCA.
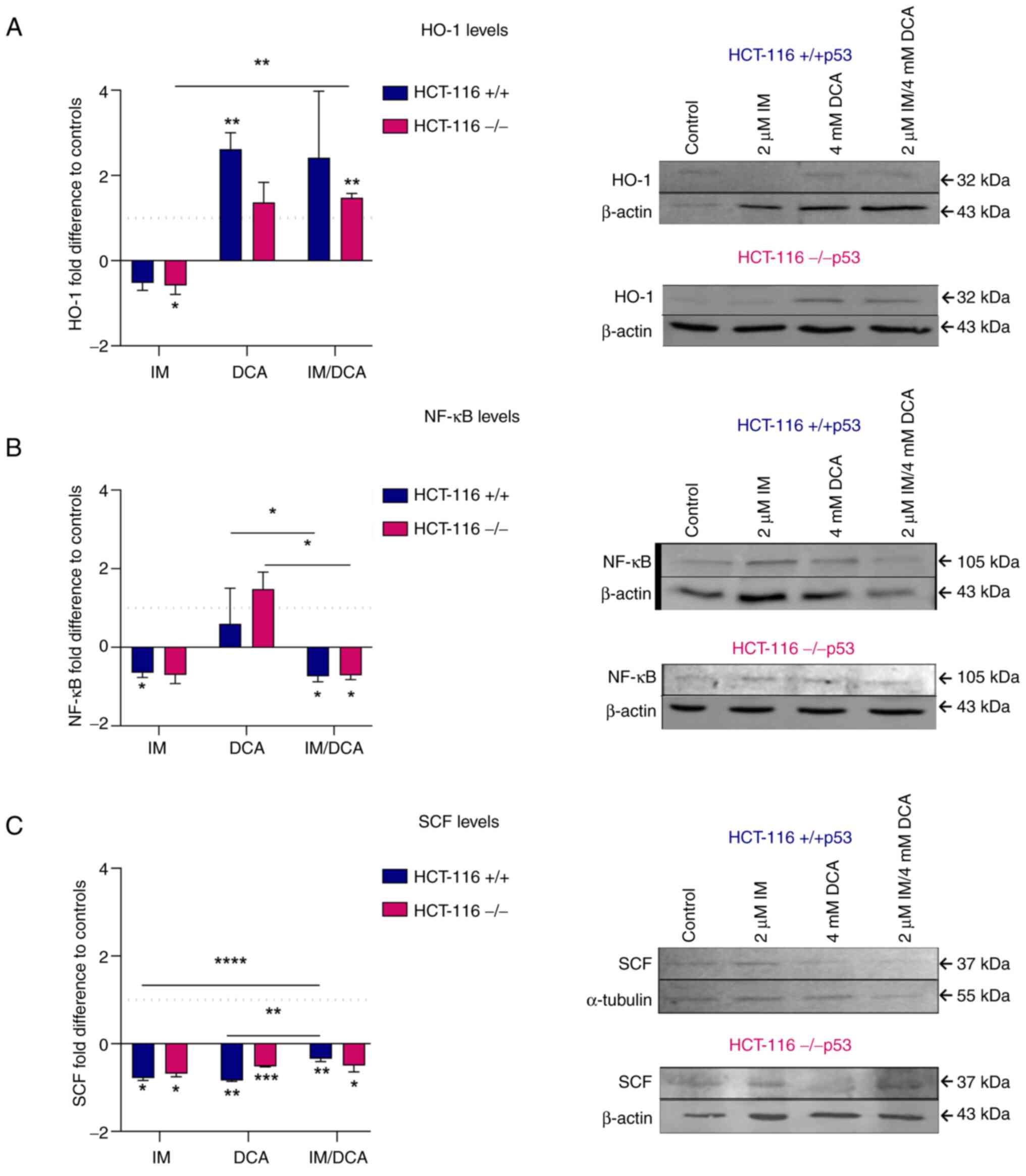
Molecular effects of IM, DCA and their combination on the levels of redox signaling mediators and hypoxia-dependent factors in HCT-116 cells
When the levels of the HO-1 protein were measured, it was noticed that treatment with IM decreased the expression levels of HO-1 marginally in both HCT-116 (+/+p53) and HCT-116 (−/−p53) cells. By contrast, treatment with DCA alone, or in combination with IM, increased the HO-1 levels in both cell types in a similar way (~2-fold) (Fig. 10A). In patients with CML, IM reduced the protein levels of NF-κB (61). In the present study, both types of HCT-116 cells, treated with IM or IM with DCA also demonstrated a mild decrease in the protein levels of NF-κB protein levels, suggesting overall that the p53/NF-κB axis is not significantly influenced by IM (Fig. 10B). On the other hand, treatment of both cell types with DCA led to a mild increase of NF-κB. It was also observed that IM could marginally decrease the protein levels of SCF in both HCT-116 cells which is in agreement with the literature, as IM has been identified to block the SCF/c-Kit pathway (42). Even in the absence of full-length p53 protein, IM decreased the proteins levels of SCF in HCT-116 (−/−p53) cells, implicating that IM probably exerts its action through a p53 independent pathway (Fig. 10C). DCA as well as the combination of DCA with IM (IM/DCA) also mildly decreased the SCF protein levels in HCT-116 cells (Fig. 10C).
Discussion
Tumor cells of diverse origins, such as the ones derived from hematological malignancies (such as leukemias) and/or solid tumors, exhibit imbalance in the dynamics of cellular metabolism and mitochondrial bioenergetics. Evidence now exists to indicate these cells have been retuned in the regulation thresholds of OxPhos and aerobic glycolysis that vary extensively among different types and populations of tumors (3,32).
A previous study by the authors (24) revealed that IM disrupts HDCBAP in Bcr-Abl+ cells by downregulating the expression of key genes. In this context, heme is essential for the biogenesis and proper functioning of mitochondrial respiratory chain complexes. Moreover, mitochondria and cancer are linked through the generation of ROS (5,8), while ROS are mediating mechanisms of action in targeted cancer therapy strategies (62).
In the present study, the levels of OxPhos related SCO2 and FXN proteins, as well as additional metabolic enzymes and hypoxic factors involved in glycolysis and oxidative stress were assessed and shown to share physicochemical interactions (Table I). The central aim of the present study was to determine the effects of IM and DCA on mitochondrial bioenergetics of two cancer cell lines. It is conceivable that all these macromolecules interact within the mitochondrial bioenergetic playground as demonstrated in Fig. S5A and B.
Metabolic enzymes involved in glycolysis include PKM2, GAPDH and LDHA. In addition, the hypoxic tumor cells often express relatively high levels of HIF-1a, SCF, HO-1 and NF-κB. Taken together, these aspects indicated that transitioning of mitochondrial bioenergetics occurs within the frame of the 'Warburg effect' as discussed in the introduction (Fig. 1A and B). That is a transitional shift from OxPhos to glycolysis. Unfortunately, to date, the precise molecular mechanisms involved in this metabolic reprogramming have not been fully explored (3).
Earlier attempts to exploit the unique nature of the hypoxic microenvironment in tumor cells via the intracellular bio-activation of a new class of 'bioreductive alkylating agents' as potent chemotherapeutics, led to the development of cytotoxic antineoplastic agents by Sartorelli and his colleagues (63,64). More recently, Li et al (65) also insisted in targeting hypoxia with hypoxia-activated prodrugs. Thus, during the alternative approach to tackle this metabolic imbalance in mitochondrial cancer bioenergetics, the observations of the authors that IM downregulates remarkably the expression of both SCO2 and FXN genes were expanded. The former is involved in the biogenesis of COX, while the latter in the assembly of Fe/S clusters of the mitochondrial respiratory chain (66,67), both associated with COX deficiency (24). As proposed by Michelakis et al (31), DCA was utilized as a potential metabolic-targeting therapy for cancer capable of blocking the lactate production at the last steps of glycolysis. Thus, it was investigated whether these two agents, IM and DCA, which block OxPhos and glycolysis, respectively, can exhibit antineoplastic activity guiding cancer cells to death.
According to the data presented with K-562, IM provoked extensive cell death, as expected, since IM is a selective drug of choice for CML, that also downregulates SCO2 and FXN in a dose-dependent manner. The concentration of IM used in these experiments was 1-2 μM, as it corresponds to the therapeutic concentration used for CML (19,24) and revealed to be safe for patients. IM reduced the levels of both mitochondrial proteins, SCO2 and FXN in a dose-dependent fashion. Thus, IM blocked the OxPhos, in agreement with the previous results (24). Moreover, IM increased the protein levels of two glycolytic enzymes, GAPDH and LDHA, without having a significant effect on the glycolytic enzyme PKM2. An increase in the protein levels of HIF-1α, SCF and VEGF (at a markedly higher level) was also observed following treatment with IM.
DCA neither caused any substantial change in the levels of SCO2 and FXN, nor promoted any substantial alteration in GAPDH levels, as expected, as DCA inhibits the generation of lactic acid in the last step of glycolysis, directing pyruvate to OxPhos. Co-treatment of K-562 cells with IM and DCA increased the proportion of dead cells, even though slightly affected the levels of hypoxic markers HIF-1a, SCF, NF-κB and VEGF. Based on these findings, it can be inferred that in K-562 cells, it is likely that the effect of IM exceeded the one of DCA and combination treatment made these cells more sensitive to IM treatment and more susceptible to cell death. Furthermore, as these two agents target distinct cellular processes, OxPhos and glycolysis, the suppressive effects of the combination treatment on cell proliferation were considered as a cooperative effect.
It is well known that a significant percentage (10-20%) of patients with CML develop resistance to IM (Gleevec®) after administration for a certain time (68). For this reason, the effects of IM and DCA in the IM-chemoresistant K-562R cell line were investigated (resistant to IM as maintained with continuous presence of IM at 0.89 μM). IM-treated K-562R demonstrated a lower inhibition of cell proliferation, while in the presence of DCA only cell death increased. Co-treatment with both IM and DCA led to a higher percentage of cell death in K-562R cells.
Contrary to what was reported for K-562 cells, the IM-resistant K-562R cells exhibited a significant increase of SCO2, while FXN was moderately increased. These results suggested that IM-driven adaptation processes in K-562R cells increase the protein expression of a certain OxPhos component or components. Moreover, the flow cytometric analysis (Fig. 6) revealed that K-562 cells have considerably high levels of ROS and treatment with either IM, DCA or both agents markedly decreased them. K-562R cells, however, have been revealed to contain a relatively lower load of ROS, while their treatment with DCA increased ROS (Fig. 6B). As DCA alone or in combination with IM increased in the K-562R the levels of the two SCO2 and FXN mitochondrial proteins, it appeared that the DCA reversed the phenotype of resistance concerning energy production, thus promoting OxPhos. It may be possible that the increased ROS is a result from this metabolic shift towards OxPhos. Overall, K-562R exhibited milder bioener- getic changes after exposure to IM and DCA compared with the IM-sensitive K-562 cells. This could reflect a more tightly regulated redox status due to the onset of pharmacological resistance in the former CML cell line.
Regarding the glycolytic molecules studied in the K-562 lines, the expression levels of GAPDH decreased after DCA administration, as expected, while in the presence of the co-treatment, the levels of LDHA increased. Due to the interaction of HIF-1a (39) with VEGF and SCF, an increase in their levels was also observed, but to a markedly lower extent in the K-562R, and only after administration of DCA or IM/DCA. Finally, the expression protein levels of NF-κB, as in the K-562 cells, exhibited a decrease. In conclusion, in both K-562 and K-562R cells, the co-treatment of IM and DCA led to increased cytotoxicity, probably by interfering with the metabolic pathways of OxPhos and glycolysis. Notably, IM/DCA treatments revealed that only K-562R had the capacity to increase glycolytic (for example, LDHA) and OxPhos components (SCO2 and FXN) in parallel. This pattern could represent metabolomic and bioenergetic manifestations of drug resistance, similarly to what has been observed in other malignancies (69). Such findings could be valuable, especially in the therapeutics of patients with CML that develop resistance to IM.
With respect to the treatments of the HCT-116 cell lines, it was observed that IM reduced substantially the OxPhos biomarkers SCO2 and FXN in both HCT-116 cell lines. SCO2 and FXN are transcriptional targets of p53 (13). In HCT-116 (−/−p53) there is a deletion of p53 exon 2 (51,58), which is in accordance with the findings of the present study for the gene expression and protein levels, as demonstrated in Fig. S1. Treatment of HCT-116 (+/+p53) with DCA increased the SCO2 levels. Based on the study of Allende-Vega et al (70) in 2015, HCT-116 (+/+p53) cells treated with 10 mM DCA for 24 h exhibited an increase in p53. In a similar way, DCA caused an increase in FXN protein expression levels in HCT-116 (+/+p53) cells. This result reinforced the observation that the FXN gene is also a transcriptional target of p53 and an increase in p53 protein levels could be associated also with an increase in the corresponding FXN levels (59,71,72). On the other hand, IM/DCA in HCT-116 (−/−p53) cells decreased both the SCO2 and FXN protein levels, possibly due to the absence of full length TP53 protein.
IM as well as DCA reduced the GAPDH protein levels in all HCT-116 cells, indicating that these agents interfere with glucose metabolism, either as inhibitor of OxPhos or glycolysis, respectively. In HCT-116 (+/+p53) cells, DCA reduced LDHA protein levels, in agreement with the literature (28), while in HCT-116 (−/−p53) cells this effect was reversed. It appears that in the absence of full-length p53 protein that promotes OxPhos, DCA leads to an increase in LDHA protein.
DCA inhibits the pyruvate dehydrogenase (PDH) kinase (PDK) and thus releases PDH, the rate-limiting enzyme of glucose oxidation. Considering that the HCT-116 (−/−p53) cells are more likely to be glycolytic, the lack of full-length p53 protein affected the effectiveness of IM and DCA, since these cells were revealed to be less susceptible compared with HCT-116 (+/+p53) cells as previously proposed (58). The upregulation of LDHA induced by DCA in these conditions may be part of a feedback response mechanism to support energy production under anaerobic conditions and convert the generated pyruvate to lactate that may also contribute to the acidification of the hypoxic tumor cell microenvironment. This could reflect an adaptive metabolic profile capability of the solid tumor cells that may function as a defense mechanism.
IM caused a decrease in SCF levels in HCT-116 (+/+p53) cells, also in agreement with the previous results that IM blocks the SCF/c-Kit pathway (42). Moreover, DCA caused a decrease in SCF protein in both HCT-116 cell lines, although to a smaller extend in the HCT-116 (−/−p53) cells. IM also caused a decrease in the NF-κB protein, as this has been reported in patients with CML (61). In all HCT-116 cells, IM decreased SCF protein expression levels suggesting that in relation with SCF, IM probably exerts its action independently of the p53 transcriptional control.
IM also decreased the HO-1 protein levels in all HCT-116 cells. Although the inter-relationship of HO-1 with p53 is not fully understood, it was previously suggested that p53, either induces HO-1 gene expression (downstream induction) or that HO-1 is actually upstream of the TP53 gene (44). Based on the results obtained from the two cell lines, it appears that the absence of full-length p53 protein does not affect the effect of IM on HO-1.
The findings of the present study revealed that the HCT-116 cell lines suffered cell death induced by the coordinated action of both agents, although to a different extent. In comparison with the K-562 cells (Fig. 2), the HCT-116 cells were found to be less susceptible to the simultaneous targeting of OxPhos and glycolysis with IM and DCA, respectively (Fig. 7). These findings provide an alternative chemotherapeutic approach based on the metabolic peculiarities of various tumors (3). Since metabolic reprogramming has been recognized as an emerging hallmark of cancer, DCA, an inhibitor of glycolysis (that exhibits a relatively safe and effective pharmacokinetic profile) can be combined with other potent chemotherapeutic agents (29,73,74). This development has attracted a lot of attention in numerous basic and clinical investigations of tumor therapy due to its capacity to intervene in metabolic reprogramming. A recent study described a variety of methods that can be applied to investigate the mitochondrial structure and function in cancer cells, within tissue cultures as well as in animal models (75). As an expansion of the studies conducted by the authors, a future metabolic flux analysis, could provide valuable mechanistic insights.
The results highlighted that the combination of IM with DCA is effective as a combination therapy in limiting the expansion of the two malignant cell lines examined by disrupting their bioenergetic dynamics. As these agents target the dysregulated metabolic pathways of malignant cells, these findings may have a potential clinical value for use along with other existing antineoplastic agent schemes approved for different types of cancers. These findings need further investigation at the preclinical stage by utilizing murine models with transplantable tumors.
In the present study, DCA was combined for the first time with IM. The findings of the present study indicated that this combination is quite potent in eradicating human leukemic cells more extensively as compared with the more robust human solid colorectal cancer cells. Differences recorded in the extent of cell death between these two types of cancer cells may be attributed to different threshold dynamics between OxPhos and (aerobic) glycolysis in each cancer population. Such chimeric states shape the ground for pharmacological combinations against exploitable joint targets in advanced cancer chemotherapy protocols.
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
MGK contributed to investigation, methodology, validation, data curation, formal analysis, visualization and writing of the K-562 cell lines work. AAL contributed to investigation, methodology, validation, data curation, formal analysis, visualization and writing of the HCT-116 cell lines work. GIG contributed to flow cytometry data curation, visualization of network models and review. SAT contributed to analysis and data interpretation, data curation, writing and editing. ANM contributed to methodology and data curation. ISP contributed to conceptualization, review and editing. ISV contributed to resources and conceptualization. LCP contributed to main conceptualization, project supervision and administration, data analysis and curation, resources, writing, review and editing, AST contributed to conceptualization, data analysis and interpretation, writing, review and editing. MGK, AAL and LCP confirm the authenticity of all the raw data. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Abbreviations:
|
acetyl-CoA |
acetyl coenzyme A |
|
AP |
alkaline phosphatase |
|
CML |
chronic myeloid leukemia |
|
COX |
cytochrome c oxidase |
|
DCA |
dichloroacetate |
|
DCFDA |
2′,7′-dichlorofluorescein diacetate |
|
HDCBAP |
heme dependent cytochrome c oxidase biosynthesis and assembly pathway |
|
HIF-1a |
hypoxia inducing factor-1a |
|
HO-1 |
heme oxygenase-1 |
|
FXN |
frataxin |
|
GAPDH |
glyceraldehyde-3-phosphate dehydrogenase |
|
IM |
imatinib |
|
LDHA |
lactate dehydrogenase A |
|
OxPhos |
oxidative phosphorylation |
|
p53 |
p53 transcriptional regulator |
|
PDC |
pyruvate dehydrogenase complex |
|
PDK |
pyruvate dehydrogenase kinase |
|
PKM2 |
pyruvate kinase M2 |
|
ROS |
reactive oxygen species |
|
SCO2 |
synthesis of cytochrome c oxidase 2 |
|
SCF |
stem cell factor |
|
VEGF |
vascular endothelial growth factor |
|
WB |
western blotting |
Acknowledgments
The study involving the K-562 cell lines has been submitted to the Department of Biochemistry and Biotechnology, University of Thessaly, Larissa, Greece, for awarding the degree Master of Sciences to Maria Kakafika. The work involving the HCT-116 cell lines has been submitted to the School of Pharmacy, Faculty of Health Sciences of the Aristotle University of Thessaloniki, Thessaloniki, Greece, for awarding the degree Master of Sciences to Areti Lyta. The authors would like to thank Professor Bert Vogelstein (Johns Hopkins Medical School, Baltimore, USA) for his kind donation of the HCT-116 (+/+p53) and HCT-116 (−/−p53) cells and Professor Diamanto Lazari (School of Pharmacy, Faculty of Health Sciences, Aristotle University of Thessaloniki, Thessaloniki, Greece) for her kind donation of the K-562R IM-chemoresistant cell line. Moreover, the authors would like to thank Dr Marina Gerousi (Institute of Applied Biosciences, Centre for Research and Technology Hellas, Thessaloniki, Greece) for her technical expertise with the flow cytometric analysis.
Funding
The present study was supported by internal departmental university funds from the Department of Biochemistry and Biotechnology of the University of Thessaly, Larissa, Greece and the School of Pharmacy of the Aristotle University of Thessaloniki, Thessaloniki, Greece, allocated to the Laboratory of Pharmacology for M.Sc., graduate studies.
References
|
Hanahan D and Weinberg RA: The hallmarks of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Hanahan D and Weinberg RA: Hallmarks of cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Bartman CR, Weilandt DR, Shen Y, Lee WD, Han Y, TeSlaa T, Jankowski CSR, Samarah L, Park NR, da Silva-Diz V, et al: Slow TCA flux and ATP production in primary solid tumours but not metastases. Nature. 614:349–357. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Schwartz L, Supuran CT and Alfarouk KO: The warburg effect and the hallmarks of cancer. Anticancer Agents Med Chem. 17:164–170. 2017. View Article : Google Scholar | |
|
Alam MM, Lal S, FitzGerald KE and Zhang L: A holistic view of cancer bioenergetics: Mitochondrial function and respiration play fundamental roles in the development and progression of diverse tumors. Clin Transl Med. 5:32016. View Article : Google Scholar : PubMed/NCBI | |
|
Bernard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T and Rossignol R: Mitochondrial bioenergetics and structural network organization. J Cell Sci. 120:838–848. 2007. View Article : Google Scholar | |
|
Blasiak J, Hoser G, Bialkowska-Warzecha J, Pawlowska E and Skorski T: Reactive oxygen species and mitochondrial DNA damage and repair in BCR-ABL1 cells resistant to imatinib. Biores Open Access. 4:334–342. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Prieto-Bermejo R, Romo-González M, Pérez-Fernández A, Ijurko C and Hernández-Hernández Á: Reactive oxygen species in haematopoiesis: Leukaemic cells take a walk on the wild side. J Exp Clin Cancer Res. 37:1–18. 2018. View Article : Google Scholar | |
|
Murphy MP: How mitochondria produce reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar | |
|
Warburg O: On respiratory impairment in cancer cells. Science. 124:269–270. 1956. View Article : Google Scholar : PubMed/NCBI | |
|
Warburg O: The metabolism of carcinoma cells. J Cancer Res. 9:148–163. 1925. View Article : Google Scholar | |
|
Heiden MGV, Cantley LC and Thompson CB: Understanding the warburg effect: The metabolic requirements of cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar | |
|
Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F and Hwang PM: p53 regulates mitochondrial respiration. Science. 312:1650–1653. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Kamp WM, Wang PY and Hwang PM: TP53 mutation, mitochondria and cancer. Curr Opin Genet Dev. 38:16–22. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Papadopoulou LC, Sue MC, Davidson MM, Tanji K, Nishino I, Sadlock JE, Krishna S, Walker W, Selby J, Glerum DM, et al: Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet. 23:333–337. 1999. View Article : Google Scholar : PubMed/NCBI | |
|
Blandino G, Valenti F, Sacconi A and Di Agostino S: Wild type- and mutant p53 proteins in mitochondrial dysfunction: Emerging insights in cancer disease. Semin Cell Dev Biol. 98:105–117. 2020. View Article : Google Scholar | |
|
Miliotou AN, Foltopoulou PF, Ingendoh-Tsakmakidis A, Tsiftsoglou AS, Vizirianakis IS, Pappas IS and Papadopoulou LC: Protein transduction Domain-mediated delivery of recombinant proteins and in vitro transcribed mRNAs for protein replacement therapy of human severe genetic mitochondrial disorders: The case of Sco2 deficiency. Pharmaceutics. 15:2862023. View Article : Google Scholar : PubMed/NCBI | |
|
Chadha R, Shah R and Mani S: Analysis of reported SCO2 gene mutations affecting cytochrome c oxidase activity in various diseases. Bioinformation. 10:329–333. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Arora B, Gota V, Menon H, Sengar M, Nair R, Patial P and Banavali SD: Therapeutic drug monitoring for imatinib: Current status and Indian experience. Indian J Med Paediatr Oncol. 34:224–228. 2013. View Article : Google Scholar | |
|
Soverini S, Martinelli G, Lacobucci I and Baccarani M: Imatinib mesylate for the treatment of chronic myeloid leukemia. Expert Rev Anticancer Ther. 8:853–864. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Ramirez P and DiPersio JF: Therapy options in imatinib failures. Oncologist. 13:424–434. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Maekawa T, Ashihara E and Kimura S: The Bcr-Abl tyrosine kinase inhibitor imatinib and promising new agents against Philadelphia chromosome-positive leukemias. Int J Clin Oncol. 12:327–340. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Deininger M, Buchdunger E and Druker BJ: The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 105:2640–2653. 2005. View Article : Google Scholar | |
|
Papadopoulou LC, Kyriazou AV, Bonovolias ID and Tsiftsoglou AS: Imatinib inhibits the expression of SCO2 and FRATAXIN genes that encode mitochondrial proteins in human Bcr-Abl+ leukemia cells. Blood Cells Mol Dis. 53:84–90. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Chang SP, Shen SC, Lee WR, Yang LL and Chen YC: Imatinib mesylate induction of ROS-dependent apoptosis in melanoma B16F0 cells. J Dermatol Sci. 62:183–191. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Redza-Dutordoir M and Averill-Bates DA: Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 1863:2977–2992. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, Castoria G and Migliaccio A: ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 52:192–203. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Kon S, Ishibashi K, Katoh H, Kitamoto S, Shirai T, Tanaka S, Kajita M, Ishikawa S, Yamauchi H and Yako Y: Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol. 19:530–541. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Tataranni T and Piccoli C: Dichloroacetate (DCA) and cancer: An overview towards clinical applications. Oxid Med Cell Longev. 2019:82010792019. View Article : Google Scholar : PubMed/NCBI | |
|
Delaney LM, Ho N, Morrison J, Farias NR, Mosser DD and Coomber BL: Dichloroacetate affects proliferation but not survival of human colorectal cancer cells. Apoptosis. 20:63–74. 2015. View Article : Google Scholar | |
|
Michelakis ED, Webster L and Mackey JR: Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 99:989–994. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Castro I, Sampaio-Marques B and Ludovico P: Targeting metabolic reprogramming in acute myeloid leukemia. Cells. 8:9672019. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang JY, Zhang F, Hong CQ, Giuliano AE, Cui XJ, Zhou GJ, Zhang GJ and Cui YK: Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol Med. 12:10–22. 2015.PubMed/NCBI | |
|
Gupta V and Bamezai RNK: Human pyruvate kinase M2: A multifunctional protein. Protein Sci. 19:2031–2044. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Pouysségur J and Ždralević M: Reply to beltinger: Double genetic disruption of lactate dehydrogenases A and B is required to ablate the 'Warburg effect' restricting tumor growth to oxidative metabolism. J Biol Chem. 294:672019. View Article : Google Scholar | |
|
Beltinger C: LDHA and LDHB are dispensable for aerobic glycolysis in neuroblastoma cells while promoting their aggressiveness. J Biol Chem. 294:662019. View Article : Google Scholar : PubMed/NCBI | |
|
Ždralević M, Brand A, Di Ianni L, Dettmer K, Reinders J, Singer K, Peter K, Schnell A, Bruss C, Decking SM, et al: Double genetic disruption of lactate dehydrogenases A and B is required to ablate the 'Warburg effect' restricting tumor growth to oxidative metabolism. J Biol Chem. 293:15947–15961. 2018. View Article : Google Scholar | |
|
Valvona CJ, Fillmore HL, Nunn PB and Pilkington GJ: The regulation and function of lactate dehydrogenase A: Therapeutic potential in brain tumor. Brain Pathol. 26:3–17. 2016. View Article : Google Scholar | |
|
Missiaen R, Lesner NP and Simon MC: HIF: A master regulator of nutrient availability and metabolic cross-talk in the tumor microenvironment. EMBO J. 42:e1120672023. View Article : Google Scholar : PubMed/NCBI | |
|
Stubbs M and Griffiths JR: The altered metabolism of tumors: HIF-1 and its role in the Warburg effect. Adv Enzyme Regul. 50:44–55. 2010. View Article : Google Scholar | |
|
Gavriilidis GI, Ntoufa S, Papakostantinou N, Kotta K, Koletsa T, Chartomatsidou E, Moysiadis T, Stavroyianni N, Anagnostopoulos A, Papadaki E, et al: Stem cell factor is implicated in microenvironmental interactions and cellular dynamics of chronic lymphocytic leukemia. Haematologica. 106:692–700. 2021. View Article : Google Scholar : | |
|
Eberle F, Leinberger FH, Saulich MF, Seeger W, Engenhart-Cabillic R, Hänze J, Hattar K, Dikomey E and Subtil FSB: In cancer cell lines inhibition of SCF/c-Kit pathway leads to radiosensitization only when SCF is strongly over-expressed. Clin Transl Radiat Oncol. 2:69–75. 2017.PubMed/NCBI | |
|
Chiang SK, Chen SE and Chang LC: The role of HO-1 and its crosstalk with oxidative stress in cancer cell survival. Cells. 10:24012021. View Article : Google Scholar : PubMed/NCBI | |
|
Andrés NC, Fermento ME, Gandini NA, Romero AL, Ferro A, Donna LG, Curino AC and Facchinetti MM: Heme oxygenase-1 has antitumoral effects in colorectal cancer: Involvement of p53. Exp Mol Pathol. 97:321–331. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Lingappan K: NF-κB in oxidative stress. Curr Opin Toxicol. 7:81–86. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Johnson RF and Perkins ND: Nuclear factor-κB, p53, and mitochondria: Regulation of cellular metabolism and the Warburg effect. Trends Biochem Sci. 37:317–324. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, Moretti M, De Smaele E, Beg AA, Tergaonkar V, et al: NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol. 13:1272–1279. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Lozzio BB and Lozzio CB: Properties and usefulness of the original K-562 human myelogenous leukemia cell line. Leuk Res. 3:363–370. 1979. View Article : Google Scholar : PubMed/NCBI | |
|
Głowacki S, Synowiec E, Szwed M, Toma M, Skorski T and Śliwiński T: Relationship between oxidative stress and imatinib resistance in model chronic myeloid leukemia cells. Biomolecules. 11:6102021. View Article : Google Scholar : | |
|
Lee F, Fandi A and Voi M: Overcoming kinase resistance in chronic myeloid leukemia. Int J Biochem Cell Biol. 40:334–343. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B: Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI | |
|
Bourdon JC: p53 and its isoforms in cancer. Br J Cancer. 97:277–282. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Georgiou-Siafis SK and Tsiftsoglou AS: Activation of KEAP1/NRF2 stress signaling involved in the molecular basis of hemin-induced cytotoxicity in human pro-erythroid K562 cells. Biochem Pharmacol. 175:1139002020. View Article : Google Scholar : PubMed/NCBI | |
|
Foltopoulou PF, Tsiftsoglou AS, Bonovolias ID, Ingendoh AT and Papadopoulou LC: Intracellular delivery of full length recombinant human mitochondrial L-Sco2 protein into the mitochondria of permanent cell lines and SCO2 deficient patient's primary cells. Biochim Biophys Acta. 1802:497–508. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Bradford M: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of Protein-dye binding. Anal Biochem. 72:248–254. 1976. View Article : Google Scholar : PubMed/NCBI | |
|
Eruslanov E and Kusmartsev S: Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol. 594:57–72. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al: The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49:108002021. View Article : Google Scholar | |
|
Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, Williams J, Lengauer C, Kinzler KW and Vogelstein B: Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 104:263–269. 1999. View Article : Google Scholar : PubMed/NCBI | |
|
Shimizu R, Lan NN, Tai TT, Adachi Y, Kawazoe A, Mu A and Taketani S: P53 directly regulates the transcription of the human frataxin gene and its lack of regulation in tumor cells decreases the utilization of mitochondrial iron. Gene. 551:79–85. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Zhou Y, Niu W, Luo Y, Li H, Xie Y, Wang H, Liu Y, Fan S, Li Z, Xiong W, et al: p53/Lactate dehydrogenase A axis negatively regulates aerobic glycolysis and tumor progression in breast cancer expressing wild-type p53. Cancer Sci. 110:939–949. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Ciarcia R, Vitiello MT, Galdiero M, Pacilio C, Iovane V, d'Angelo D, Pagnini D, Caparrotti G, Conti D, Tomei V, et al: Imatinib treatment inhibit IL-6, IL-8, NF-KB and AP-1 production and modulate intracellular calcium in CML patients. J Cell Physiol. 227:2798–2803. 2012. View Article : Google Scholar | |
|
Teppo HR, Soini Y and Karihtala P: Reactive oxygen Species-mediated mechanisms of action of targeted cancer therapy. Oxid Med Cell Longev. 2017:14852832017. View Article : Google Scholar : PubMed/NCBI | |
|
Rockwell S, Sartorelli AC, Tomasz M and Kennedy KA: Cellular pharmacology of quinone bioreductive alkylating agents. Cancer Metastasis Rev. 12:165–176. 1993. View Article : Google Scholar : PubMed/NCBI | |
|
Kennedy KA, Teicher BA, Rockwell S and Sartorelli AC: The hypoxic tumor cell: A target for selective cancer chemotherapy. Biochem Pharmacol. 29:1–8. 1980. View Article : Google Scholar : PubMed/NCBI | |
|
Li Y, Zhao L and Li XF: Targeting Hypoxia: Hypoxia-activated prodrugs in cancer therapy. Front Oncol. 11:29202021. | |
|
Patraa S and Barondeaua DP: Mechanism of activation of the human cysteine desulfurase complex by frataxin. Proc Natl Acad Sci USA. 116:19421–19430. 2019. View Article : Google Scholar | |
|
Parent A, Elduque X, Cornu D, Belot L, Le Caer JP, Grandas A, Toledano MB and D'Autréaux B: Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat Commun. 6:56862015. View Article : Google Scholar : PubMed/NCBI | |
|
Grosso S, Puissant A, Dufies M, Colosetti P, Jacquel A, Lebrigand K, Barbry P, Deckert M, Cassuto JP, Mari B and Auberger P: Gene expression profiling of imatinib and PD166326-resistant CML cell lines identifies Fyn as a gene associated withresistance to BCR-ABL inhibitors. Mol Cancer Ther. 8:1924–1933. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Bosc C, Selak MA and Sarry JE: Resistance is futile: Targeting mitochondrial energetics and metabolism to overcome drug resistance in cancer treatment. Cell Metab. 26:705–707. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Allende-Vega N, Krzywi nska E, Orecchioni S, Lopez-Royuela N, Reggiani F, Talarico G, Rossi JF, Rossignol R, Hicheri Y, Cartron G, et al: The presence of wild type p53 in hematological cancers improves the efficacy of combinational therapy targeting metabolism. Oncotarget. 6:19228–19245. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Maio N, Jain A and Rouault TA: Mammalian iron-sulfur cluster biogenesis: Recent insights into the roles of frataxin, acyl carrier protein and ATPase-mediated transfer to recipient proteins. Curr Opin Chem Biol. 55:34–44. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Sawamoto M, Imai T, Umeda M, Fukuda K, Kataoka T and Taketani S: The p53-dependent expression of frataxin controls 5-aminolevulinic acid-induced accumulation of protoporphyrin IX and photo-damage in cancerous cells. Photochem Photobiol. 89:163–172. 2013. View Article : Google Scholar | |
|
Parczyk J, Ruhnau J, Pelz C, Schilling M, Wu H, Piaskowski NN, Eickholt B, Kühn H, Danker K and Klein A: Dichloroacetate and PX-478 exhibit strong synergistic effects in a various number of cancer cell lines. BMC Cancer. 21:4812021. View Article : Google Scholar : PubMed/NCBI | |
|
Sanchez WY, McGee SL, Connor T, Mottram B, Wilkinson A, Whitehead JP, Vuckovic S and Catley L: Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br J Cancer. 108:1624–1633. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Rickard BP, Overchuk M, Chappell VA, Kemal Ruhi M, Sinawang PD, Nguyen Hoang TT, Akin D, Demirci U, Franco W, Fenton SE, et al: Methods to evaluate changes in mitochondrial structure and function in cancer. Cancers (Basel). 15:25642023. View Article : Google Scholar : PubMed/NCBI |