Chimeric antigen receptor T cells in the treatment of osteosarcoma (Review)
- Authors:
- Published online on: February 22, 2024 https://doi.org/10.3892/ijo.2024.5628
- Article Number: 40
-
Copyright: © Yu et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
1. Introduction
Osteosarcoma (OS) is a frequently occurring primary bone tumor in children, adolescents and young adults with a worldwide incidence rate of 3.4 cases per 10,000 individuals per year (1,2). OS commonly occurs in the metaphysis of long bones, mostly in the humerus, distal femur and proximal tibia, and is characterized by swelling and persistent pain in the affected bone (3). Although the combination of radical surgery and systemic chemotherapy improves the prognosis of patients with primary OS, its side effects significantly affect patients' quality of life. Moreover, the prognosis of metastatic or recurrent OS remains unsatisfactory, as the recurrence and/or metastasis rate of OS is >30%. Additionally, in certain sarcomas such as early stage uterine leiomyosarcoma, adjuvant chemotherapy (with or without radiotherapy) has shown no significant decrease in recurrence rates (4). The resistance of tumor cells to both radiotherapy and chemotherapy leads to a 5-year overall survival rate of <25% in patients with metastatic or recurrent OS (5). Rizzo et al (6) suggested that a more in-depth biological characterization was essential to comprehend the molecular biology of sarcomas and to improve identification of patients who could benefit from adjuvant therapy. Immunotherapies such as immune checkpoint inhibitors have been developed as groundbreaking treatments for patients with cancer (7). More recently, chimeric antigen receptor T (CAR-T) cell therapy, which is known as an innovative form of immunotherapy, has garnered significant attention.
In 1993, Stancovski et al (8) made progress in the improvement of CAR-T cells. Nevertheless, the first generation of CAR-T cells used in initial clinical trials demonstrated limited survival rates in patients with cancer and a weak antitumor response. In 2013, Grupp et al (9) first used CD19-CAR-T cells to treat children with acute lymphoblastic leukemia (ALL). During treatment, one child experienced serious complications, but the disease was eventually steadily alleviated. In 2017, the USA Food and Drug Administration (FDA) approved two CAR-T cells targeting the CD19 protein, named Kymriah and Yescarta, for the treatment of ALL and diffuse large B-cell lymphoma, respectively, and ~83% of patients treated with Kymriah achieved a response of ≤5 years after 3 months of treatment (10,11). In 2020, Astolfi et al (12) published a study indicating that the inactivation of tumor suppressor genes was the primary driver of sarcomas. The authors suggested that future therapeutic strategies should focus on targeting the key genetic drivers of sarcoma oncogenesis. Several clinical trials have been conducted to assess the efficacy of CAR-T cell therapy for the treatment of multiple myeloma, exploring various types of CAR-T cells, such as B-cell maturation antigen CAR-T cells (13-15), CD19-CAR-T cells (16) and CD138-CAR-T cells (17). The results have revealed favorable outcomes, with CAR-T cell therapy demonstrating significant inhibition of multiple myeloma, and the overall response rates ranging from 60 to 92% (13-17).
CAR-T cell therapy has become a groundbreaking treatment for hematological malignancies and multiple myeloma. However, recent research has focused on extending the use of CAR-T therapy to treat OS, which is a solid tumor and different from blood tumors. Although CAR-T cells have excellent effectiveness in hematological tumors and have been approved for use in hematological tumors, their use in the treatment of OS remains in its infancy, and there are still problems such as difficult homing and colonization of CAR-T cells, off-target effects, immune escape, immunosuppressive tumor microenvironment (TME) and cytokine release syndrome (CRS) (18). To address these issues, previous studies have tried a variety of CAR-T cells to treat OS and have obtained favorable results, for example, increased elimination of OS and reduced incidence of adverse effects (19-23).
The present study introduced in detail the structure of CAR-T cells and the influence of changing their structure on the therapeutic effect of CAR-T cells, which should help physicians to design CAR-T cells for optimal therapeutic effect (19,20). In addition, the current study provided an overview of all target antigens that can be used to treat OS and the results of the corresponding CAR-T cells (21-31). Compared with other reviews on CAR-T cell therapy for OS published in the literature, the present review described in detail the challenges that CAR-T cell therapy faces (32,33), the ways to solve these problems (34-41), and the advantages of combining radiotherapy (42), chemotherapy (43) and chemoradiotherapy (44), so that bone oncologists can have a more comprehensive understanding of CAR-T cell for OS.
2. Structure of CAR-T cells
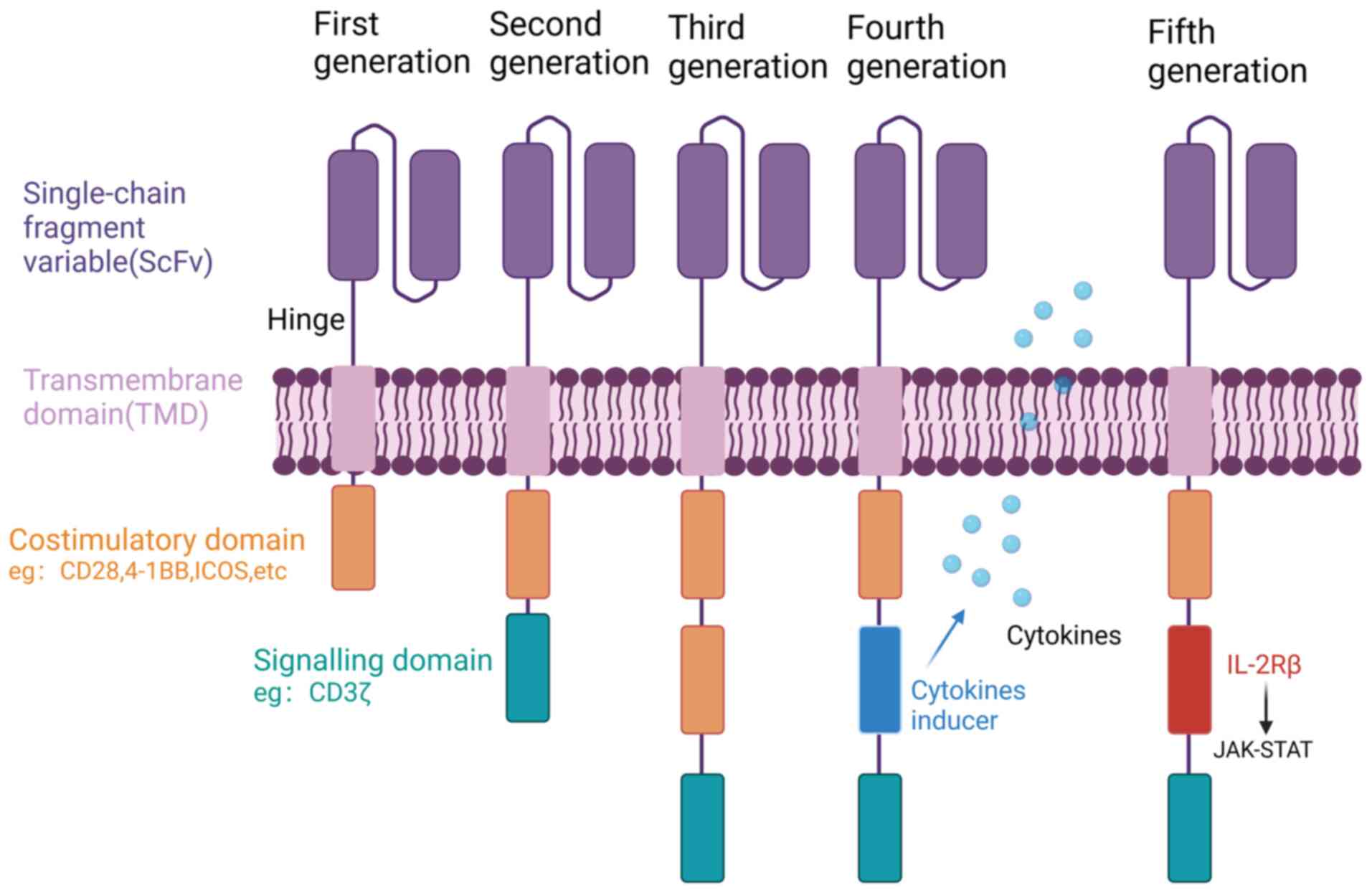
CAR-T cell therapy is a novel form of immunotherapy that utilizes genetic engineering techniques to modify and boost a patient's own immune cells outside the body (45). The therapy involves the use of a genetically engineered chimeric receptor called CAR, which is composed of various components, including an extracellular single-chain fragment variable (scFv), a hinge domain (HD), a transmembrane domain (TMD) and an intracellular domain (Fig. 1). The scFv is responsible for recognizing specific antigens on the surface of tumor cells, while the HD serves as a bridge between the scFv and TMD. The TMD anchors CAR to the membranes of T cells and promotes signal transduction into the cells, while the intracellular domain is responsible for T cell activation (19). First-generation CARs comprise a scFv and an intracellular CD3ζ activation domain. However, they have inadequate antitumor activity and proliferative capacity due to the absence of costimulatory signaling. To address this limitation, second-generation CARs have been developed. These incorporate an additional costimulatory domain, for example CD28, 4-1BB, OX40 or inducible costimulator (ICOS), which enhances their ability to proliferate and release a greater number of cytokines. At present, the marketable CAR-T cell products predominantly employ second-generation CAR structures. However, there have been advancements in the development of third-generation CARs, which incorporate two different costimulatory molecules, including CD28 and 4-1BB, to enhance their efficacy (46). Furthermore, fourth-generation CARs have emerged, which besides costimulatory domains, contain domains that regulate cytokine release. These cytokines, including interleukin (IL)-12, IL-18, IL-21 and IL-23, have the function of improving the TME and promoting the production of memory T cells, leading to increased persistence of CAR-T cells in vivo (47). Fifth-generation CARs incorporate IL-2Rβ membrane receptors, which offer a docking site for signal transducer and activator of transcription 3 (STAT3) and trigger the activation of a Janus kinase (JAK)-STAT signaling domain. Additionally, these advanced CARs exhibit a combined activation of triple signals originating from the CD3ζ, costimulatory domain and the cytokine-inducing JAK-STAT3/5 pathway (48). CARs and T cells are assembled by lentiviral or other vectors to form CAR-T cells. When CAR-T cells specifically recognize the target antigen, the T cells are activated via the intracellular STD and play a tumor cell-eliminating role (45).
Traditional immunotherapies include monoclonal antibodies, tumor vaccines, oncolytic viruses, immune checkpoint inhibitors, and adoptive transfer of activated T cells and natural killer (NK) cells in vitro (49). Upon contact with tumor antigens, B cells produce highly homogeneous monoclonal antibodies that target only specific antigenic epitopes. Binding of the monoclonal antibodies to their target antigen can activate the complement system to promote the triggering of antibody-dependent cytotoxicity (50,51). Compared with monoclonal antibodies, CAR-T cells possess numerous advantages: Firstly, CAR-T cells can recognize tumor cells even when the expression of the targeted antigen is low, whereas monoclonal antibodies cannot do so; secondly, CAR-T cells are not only able to eliminate tumor cells directly, but also secrete cytokines such as IFN-γ, which enhance their antitumor effect; and thirdly, CAR-T cells are able to proliferate and maintain a long-lasting antitumor effect after infusion (52,53).
3. Antigen targets
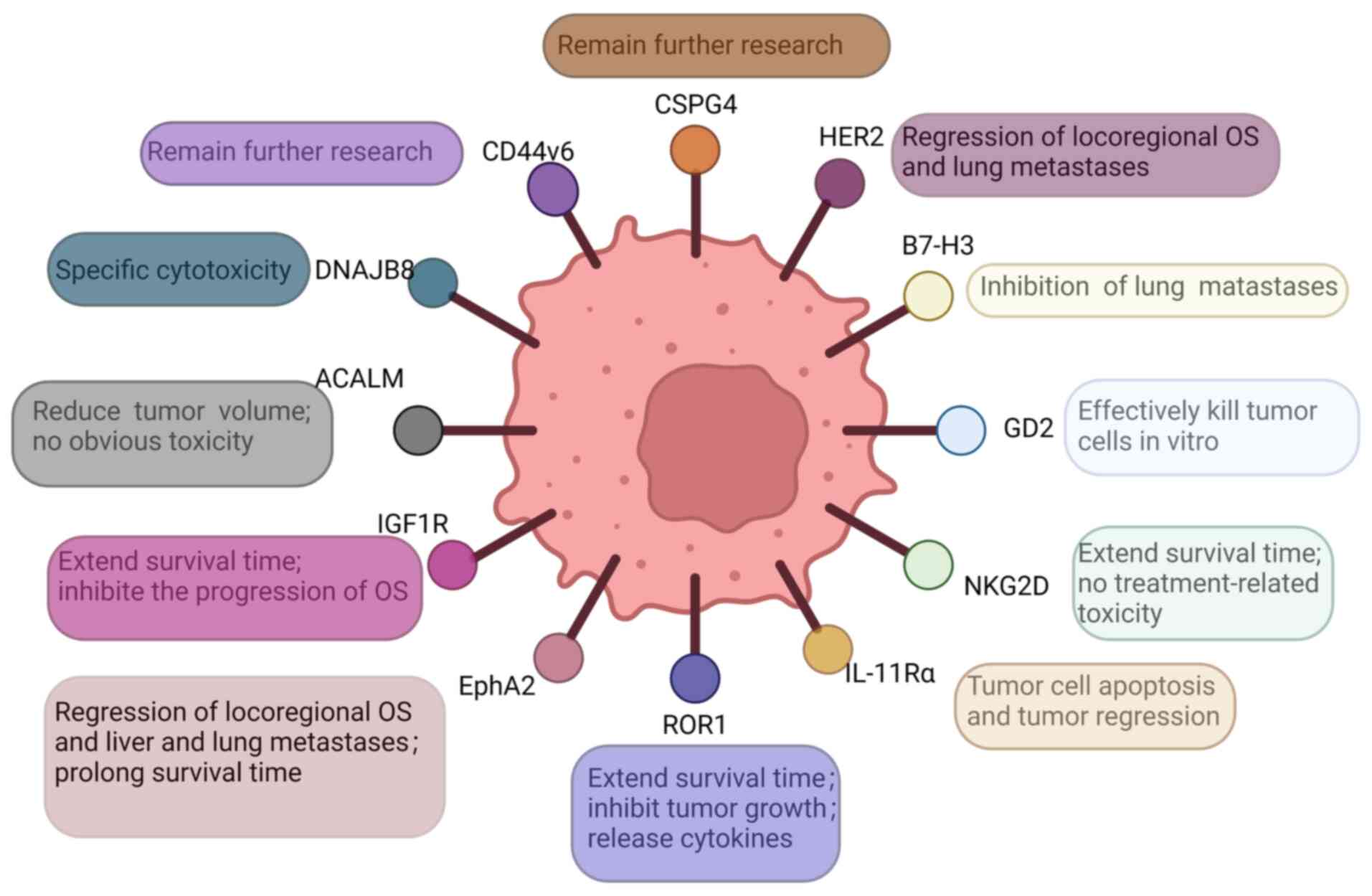
Several target antigens for the treatment of OS have been reported. In the following sections, some of these antigens are presented, including DnaJ homolog subfamily B member 8 (DNAJB8), human epidermal growth factor receptor 2 (HER2), CD276, activated leukocyte cell adhesion molecule (ALCAM, also known as CD166), EphA2, GD2, IL-11 receptor α chain (IL-11Rα), insulin-like growth factor type I receptor (IGF-1R), receptor tyrosine kinase-like orphan receptor 1 (ROR1), NK group 2D (NKG2D), chondroitin sulfate proteoglycan 4 (CSPG4) and CD44v6 (Fig. 2).
DNAJB8
DNAJB8 belongs to the heat shock protein 40 family. In health tissues, DNAJB8 is expressed only in the testis. In tumor tissues, DNAJB8 is preferentially expressed in cancer stem cells and has the function of cancer stem cell maintenance. Cancer stem cells can promote tumorigenesis, self-renewal and differentiation. Previous studies found that the overexpression of DNAJB8 increased the percentage of cells in the population representing cancer stem cells in renal cell carcinoma, and enhanced their ability to initiate tumors (21,54,55). Morita et al (21) proposed that DNAJB8-specific cytotoxic T lymphocytes had higher eliminating activity against colorectal cancer stem cells and exhibited antitumor effects. Watanabe et al (56) successfully developed second-generation CARs targeting a peptide complex derived from DNAJB8, and confirmed that DNAJB8-CAR-T cells showed a target eliminating effect on OS cells, indicating that DNAJB8 could serve as a potential and promising target antigen of CAR-T cell for OS.
HER2
HER 2 is a member of the epidermal growth factor receptor family, and plays an important role in cell proliferation (57). HER 2 is unique among the epidermal growth factor receptor family members due to its role in regulating cell proliferation and differentiation. Abnormalities in HER2 expression or function have been implicated in various malignancies, such as OS, where HER2 overexpression is associated with aggressive tumor growth. In the majority of OS cases, HER2 positivity is observed regardless of histological grade, and Abdou et al (58) indicated that positive expression of HER2 was significantly correlated with high-grade OS. Ahmed et al (52) demonstrated that HER2-CAR-T cells could eliminate HER2+ OS cells. A phase I/II clinical trial by Ahmed et al (22) revealed that patients with cancer could be transfused with safe doses of HER2-CAR-T cells that could reach the tumor and maintain it at low levels for >6 weeks in a dose-dependent manner. Thus, according to the aforementioned results, HER2 has been demonstrated to be an effective target antigen in CAR T-cell therapy for OS.
CD276
CD276 is an important immune checkpoint molecule belonging to the B7 family. It plays an essential role in modulating immune responses by exerting inhibitory effects on the activation, proliferation and cytokine production of T cells. Overexpression of CD276 is commonly observed in various cancer types, including OS, melanoma, Ewing sarcoma, non-small cell lung cancer and rhabdomyosarcoma (23). Picarda et al (23) reported that 91.8% of tumor cells in OS tissues expressed CD276, whereas the expression of CD276 in normal tissues was low. Accordingly, CD276 may be a potential target for the treatment of OS. CD276-CAR-T cells induced the regression of esophageal cancer and prolonged the survival time of mice, in addition to showing significant antitumor effects in radiotherapy-resistant prostate adenocarcinoma, ovarian cancer, pancreatic adenocarcinoma, atypical teratoid/rhabdoid tumor and neuroblastoma models (59-62). Previous studies have reported that CD276-CAR-T cells exhibit a dose-dependent antitumor activity and inhibit the development of spontaneous lung metastases in a mouse model of OS (63,64). Since CD276 has T-cell inhibitory effects, anti-CD276 CAR-T cells are a promising immunotherapy for the treatment of OS.
CD166
CD166 belongs to the immunoglobulin superfamily and plays a role in a variety of biological activities, including neuronal growth, hematopoiesis and inflammatory responses (65). CD166 has been found in primary OS biopsy specimens, where its expression level is high (66). Flow cytometric analysis indicated that the expression of CD166 on OS cell lines varied and ranged from 36.9 to 96.7%, which suggested that CD166-CAR-T cells could be a promising treatment approach for OS (24). CD166-CAR-T cells exhibit complete activation upon encountering CD166+ OS cells, effectively eliminating OS cell lines in vitro. Encouragingly, when CD166-CAR-T cells were infused into mice, there was a notable regression of tumors without causing significant toxicity (24). This promising outcome highlights the potential of CD166-CAR-T cell therapy as a safe and effective method for treating OS. In addition, CD166-CAR-T cells showed strong cytotoxicity against colorectal cancer stem cells in vitro, thus they may be an effective method for the clinical treatment of colorectal cancer (67). According to the aforementioned studies, CD166 appears to be an essential target antigen in the treatment of OS.
EphA2
During embryonic development, EphA2 serves as a tyrosine kinase receptor for Eph signaling. However, in the later stages, EphA2 expression becomes predominantly limited to specific epithelial cells (68). Overexpression of EphA2 has been frequently detected in various cancer types, including OS, breast cancer, ovarian cancer and prostate adenocarcinoma (69,70). Compared with those in normal bone tissue, EphA2 protein levels were significantly elevated in OS bone tissue. Fritsche-Guenther et al (71) conducted a study where a correlation between the overexpression of EphA2 and the development of OS was observed. In an animal model of implanted OS, EphA2-CAR-T cells successfully eliminated EPHA2-overexpressing OS cells in immunodeficient mice and significantly prolonged mouse survival. Moreover, in animal models of metastatic OS, intravenously injected EphA2-CAR-T cells could target the tumors, and effectively eliminated OS liver and lung metastases (25). Based on the findings from the aforementioned studies, it can be concluded that EphA2 holds great potential as an effective and crucial target antigen for OS.
GD2
Gangliosides are glycated lipid molecules belonging to the sphingolipid group, which have the function of mediating tumor cell adhesion to extracellular matrix proteins and in directing signaling during cell death. However, their role in tumorigenesis remains unknown (72). GD2 expression has been observed to vary in tumor tissues among patients with sarcoma, central nervous system tumors and lung cancer (73). GD2-CAR-T cells have been revealed to exhibit antigen-dependent cytotoxicity against GD2-expressing lung cancer models both in vitro and in vivo (26). Long et al (74) found that third-generation GD2-CAR-T cells exhibited significant effectiveness in eliminating GD2+ OS cell lines in laboratory conditions, but showed limited efficacy in eliminating OS in vivo. Nevertheless, the phase I clinical trial (NCT02107963) has demonstrated the safety of increasing doses of third-generation GD2-CAR-T cells in patients with GD2+ OS (75), and bispecific antibodies targeting GD2 and HER2 have potent antitumor effects against OS in vitro and in vivo (76). According to the aforementioned studies, it can be concluded that GD2 holds promise as a valuable and effective target antigen for OS.
IL-11Rα
Previous studies have demonstrated that the expression of IL-11Rα on the cell surface is linked to unfavorable patient prognosis. IL-11Rα is activated by its ligand IL-11, which can activate multiple signaling cascades in its target cells and is considered an important regulator of bone homeostasis. IL-11 and IL-11R are both expressed in OS in situ and in lung metastases, and are markers of high-risk OS, while the expression of IL-11Rα is absent in neighboring health lung tissue (77,78). IL-11Rα-CAR-T cells are able to effectively eliminate OS cells in vitro, and have been observed to specifically target and accumulate in OS lung metastases following intravenous administration, while demonstrating no accumulation in the surrounding health lung tissue. The utilization of IL-11Rα-CAR-T cells for the treatment of OS led to notable apoptosis and regression of tumors in mice with lung metastases (27). These findings suggested that IL-11Rα serves as a promising, effective and crucial target antigen for OS.
IGF-1R
IGF-1 is a hormone with an insulin-like molecular structure, and overexpression of IGF-1 and its receptor (IGF-1R) has been associated with cancer development. At OS, elevated levels of IGF-1 and IGF-1R contribute to cancer development by promoting transformation, proliferation and metastasis, and by reducing susceptibility to apoptosis. Overexpression of IGF-1/IGF-1R also contributes to tumor cell survival and resistance to chemotherapy (79). It has been demonstrated that chemotherapy-resistant OS cell lines and primary OS tissues exhibit high expression of IGF-1R (80). Moreover, the application of IGF1R-CAR-T cells has been found to effectively impede the advancement of both systemic and localized OS in immunodeficient mice, leading to a significant extension in animal survival time (28). MicroRNA (miRNA or miR)-26a, miR-100 and miRNA-133a inhibit OS cell proliferation, migration and invasion, and increase tumor cell sensitivity to chemotherapy by targeting IGF1-R (81-83). In accordance with the results of the aforementioned studies, IGF1-R is a potential, effective and important target antigen for OS.
ROR1
ROR1, a transmembrane tyrosine kinase receptor, holds great importance in facilitating cancer cell migration, invasion and metastasis. Notably, elevated levels of ROR1 have been detected in both hematological malignancies and solid tumors, such as OS, Ewing sarcoma and rhabdomyosarcoma. By contrast, ROR1 is not expressed in normal adult tissues (28,84). Previous studies have shown that anti-ROR1 monoclonal antibodies significantly block the metastasis of OS cells, and ROR1-CAR-T cells exhibit specific cytotoxicity against ROR1+ OS tissues and release sarcoma-specific cytokines such as interferon (IFN)-γ, tumor necrosis factor (TNF)-α and IL-13 in vitro, while significantly inhibiting local and disseminated sarcoma growth. Furthermore, the administration of ROR1-CAR-T cells has demonstrated promising results in extending the survival time of local sarcoma models, suggesting that ROR1-CAR-T cell therapy holds potential as a management option for high-risk sarcomas (28,85). Consequently, CAR-T cells engineered by anti-ROR1 antigens are an effective measure for the treatment of OS.
NKG2D
NKG2D is a receptor responsible for activating NK cells. NKG2D ligand (NKG2DL) is expressed in various immunosuppressive cells present in the TME. This expression of NKG2DL enables tumor cells to evade immune surveillance by evading detection and elimination by NK and T cells. It has been reported that the binding of NKG2DL on tumor cells to NKG2D in NK cells could lead to cell-mediated cytotoxicity and destruction of target cells (29,86,87). NKG2D-CAR-T cell therapy has been investigated in phase II clinical trials (NCT02203825) for the treatment of hematological malignancies, and has shown promising results with no major safety concerns raised among the participants (88). Moreover, NKG2D-CAR-T cells exhibited an NKG2D-dependent mechanism to target solid tumors, including OS, and stomach, liver and cervical cancer, and demonstrated a potent antitumor activity (89-91). Fernández et al (29) indicated that NKG2D-CAR-T cells had a strong anti-OS activity. Thus, NKG2D holds great potential as an effective target antigen for OS.
CSPG4
CSPG4 is a cell surface proteoglycan that is under-expressed in healthy tissues but overexpressed in tumor cells and TME. It is involved in tumor progression by regulating intracellular signaling pathways related to tumor cell adhesion and migration. Several studies have reported that CSPG4 mediates multidrug resistance by stimulating the PI3K/AKT signaling pathway, which plays a critical role in promoting tumor cell proliferation and survival (30,92,93). The results of those studies identified that overexpression of CSPG4 was associated with shorter survival in patients with OS. Anti-CSPG4 monoclonal antibodies significantly inhibited the proliferation and migration of CSPG4+ OS cells and improved the efficacy of doxorubicin (30). Considering the results of the aforementioned studies, CSPG4 appears to be a potential, effective and important target antigen of CAR-T cells in the treatment of OS. However, clinical trials should be carried out to further assess their effectiveness.
CD44v6
CD44 is a transmembrane glycoprotein that can be expressed in its standard form (CD44s) and a variety of variants (CD44v). One of these variants is CD44v6, which regulates the extracellular matrix and inhibits tumor cell apoptosis (94,95). CD44v6 is expressed in several malignancies, including colorectal and breast cancer (96,97). A previous meta-analysis showed that overexpression of CD44v6 was associated with lower survival and metastasis in patients with OS (31). Nakajima et al (98) found that CD44v6 could be expressed in metastatic OS and was a protein of bone tumors. Although there are no studies in the literature focused on treating OS by targeting CD44v6, based on the outcomes of the aforementioned studies, CD44v6 appears to be a promising potential OS target antigen.
4. Challenges that CAR-T cell therapy faces
At present, CAR-T cell therapy for OS encounters several challenges, including off-target effect, inhibitory TME, CRS, low efficiency of CAR-T cells and heterogeneity of OS (Fig. 3).
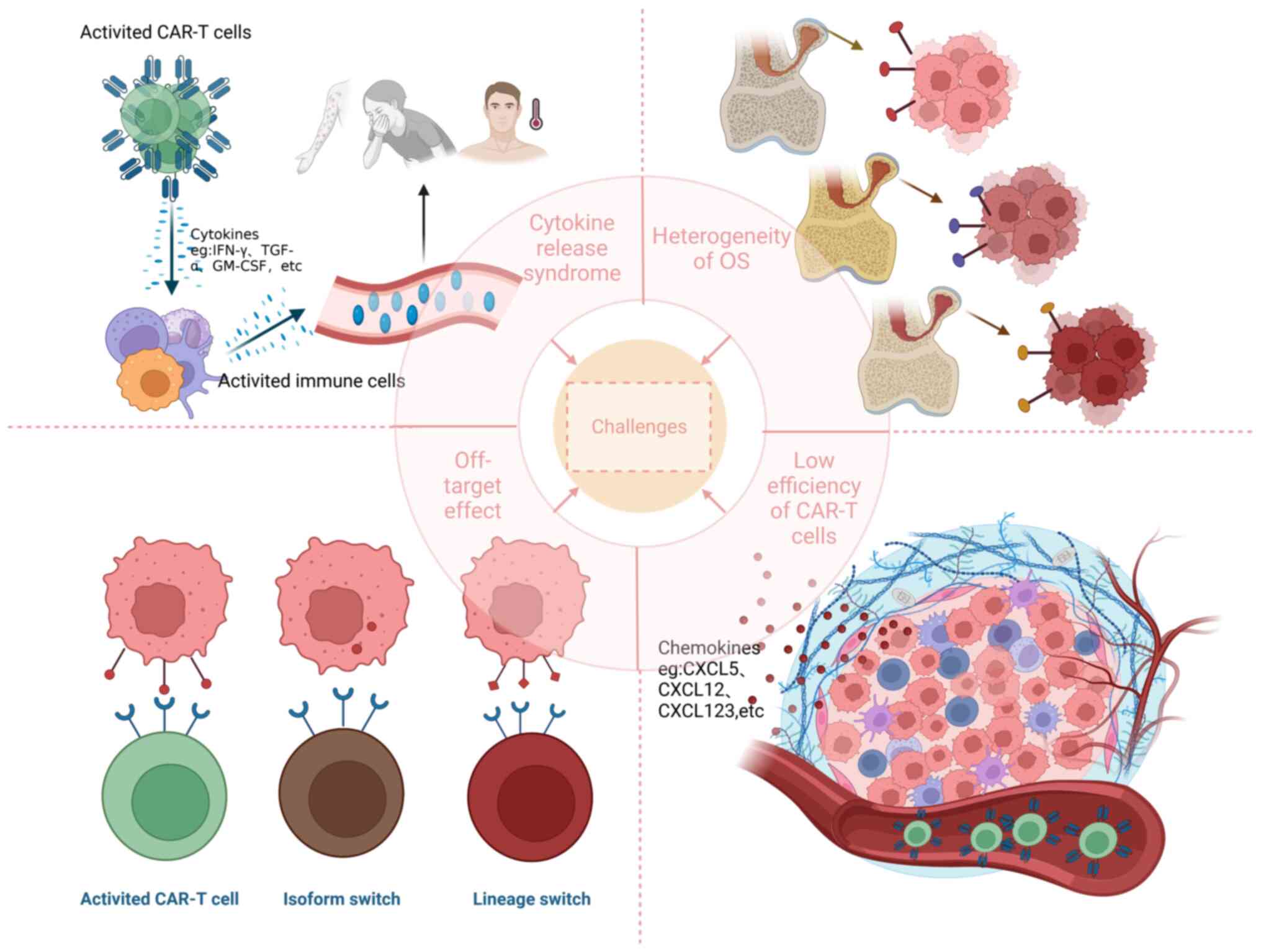
Off-target effect
Tumor antigens can be classified into two types: i) Tumor-specific antigens (TSAs) and ii) tumor-associated antigens (TAAs) (99). TSAs are unique to cancer cells and are not found in normal healthy cells, which are ideal targets for immunotherapy, as they can be specifically targeted by the immune system without affecting normal cells. However, the expression of TSAs on the surface of OS cells is quite rare, which poses challenges in developing CAR-T cells that target these specific antigens. On the other hand, TAAs are present on both cancer cells and some healthy cells, but they are overexpressed in tumors. While they may not be as specific as TSAs, they still provide viable targets for immunotherapy. Nevertheless, the disadvantage of such CAR-T cells is that they can attack normal tissue cells and therefore have certain toxic side effects (100).
For instance, CD166 is not exclusively present in OS tissues, but is also found in certain normal tissues, including epithelial cells, smooth muscle cells and neurons (101,102). A patient with colorectal cancer who received numerous third-generation CAR-T cells targeting HER2 developed respiratory distress and cardiac arrest shortly after the infusion of the T cells, and succumbed to multiple organ failure 5 days later. It was therefore hypothesized that CAR-T cells recognize HER2 in the lung epithelium despite being expressed at low levels, causing pulmonary edema and cytokine storms that lead to adverse outcomes. This was the first fatal adverse event caused by CARs recognition of target antigens outside tumor tissues (57). Thus, it is essential to carefully evaluate the safety and effectiveness of CAR-T cell therapy.
Target antigens such as DNAJB8, HER2 and CD276 have been found on the surface of OS cells, and CAR-T cells targeting these antigens have been developed to specifically eliminate tumors (22,56,103). If the antigen of the tumor cell is lost or the density of the tumor antigen is reduced, the tumor cells cannot be targeted by CAR-T cells, and therefore the tumor cannot be effectively eradicated. Based on extensive clinical experience with CD19-CAR-T cells in acute B-lymphoblastic leukemia, the loss of CD19 antigen was postulated to occur through two distinct mechanisms: i) Isoform switching and ii) lineage switching. Isoform switching refers to the fact that mutated leukemia cells lack the epitope recognized by CD19-CAR-T cells, and/or CD19 is preferentially retained in the cell and therefore not recognized by T cells, while lineage switching refers to a phenomenon in the development of myeloid leukemia where tumor cells do not express CD19 in the presence of CD19-CARs (104). The same occurs with OS. Hsu et al (25) reported an increase in tumor cells lacking EphA2 expression following EphA2-CAR-T cell therapy for OS. Consequently, immune escape may seriously affect the antitumor effect of CAR-T cells.
Inhibitory TME
CAR-T cells often encounter an inhibitory TME when infused into patients. This microenvironment can support angiogenesis, promote tumor progression and immune escape, and reduce the ability of CAR-T cells to proliferate and persist in vivo (Fig. 4) (105,106). The following subsection introduces specific immunosuppressive mechanisms.
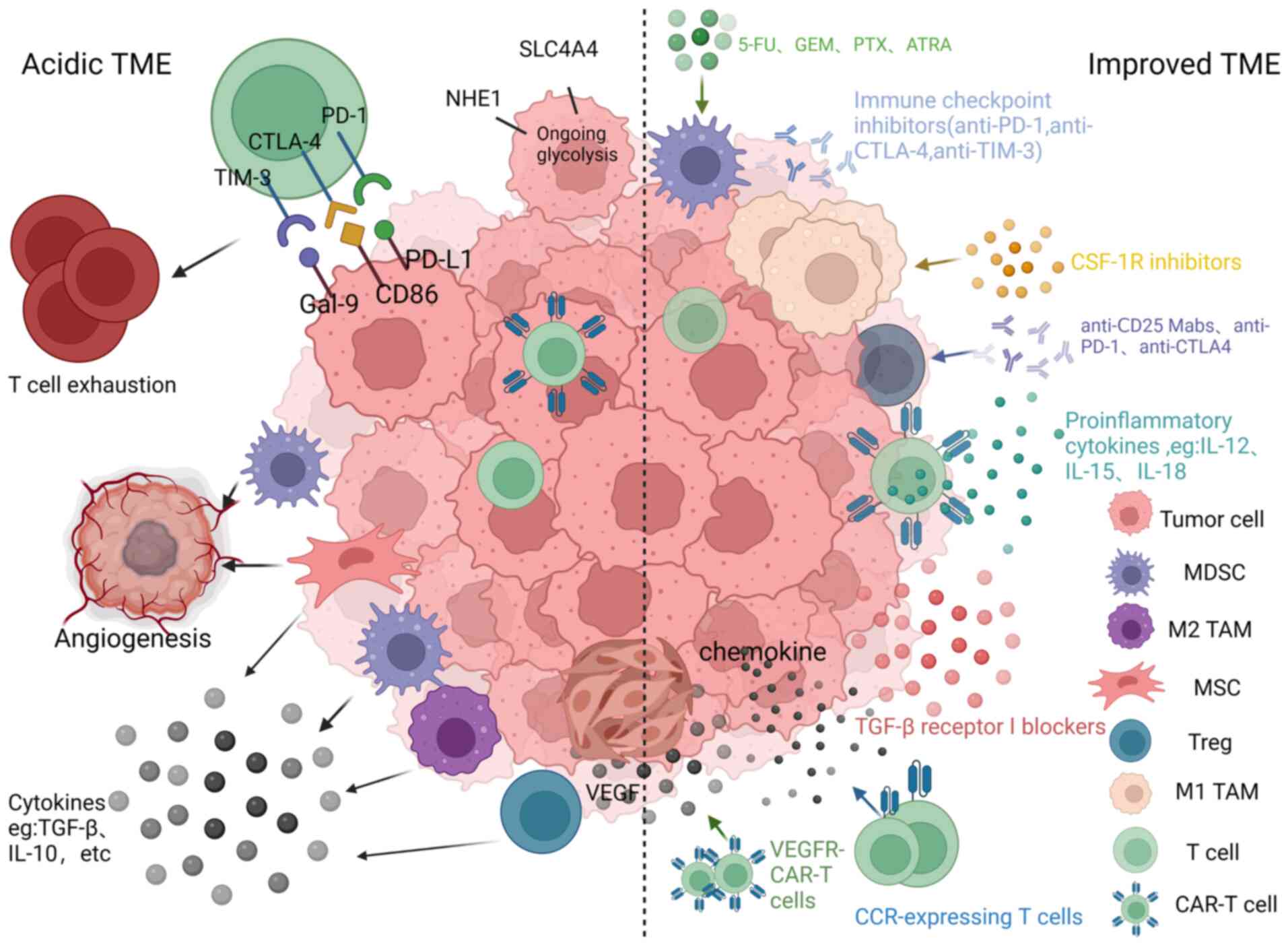
Immune checkpoints
Immune checkpoints serve as immunosuppressive molecules that help to regulate the intensity and magnitude of the immune response. Immune checkpoints, including cytotoxic T lymphocyte antigen 4 (CTLA-4), TIM-3, Gal-9, programmed death receptor 1 (PD-1) and PD ligand 1 (PD-L1), play a crucial role in promoting immune tolerance during tumor formation and development (107). PD-1 is expressed on activated effector T cells and other tumor-infiltrating lymphocytes (TILs), including NK cells. Its presence inhibits immune responses in activated T cells, resulting in a suppressed immune response. This phenomenon, namely the fact that PD-1+ T cells are no longer cytotoxic and therefore cannot fight tumors, is called T cell exhaustion (108). Hashimoto et al (109) studied 16 samples of OS, and suggested that PD-1 and PD-L1 may be associated with tumor recurrence, metastasis and patient mortality. CTLA-4 was found to be expressed in OS cells. Administration of anti-CTLA-4 antibodies has been observed to augment the antitumor response of cytotoxic T cells (110,111). In addition, TIM-3 and Gal-9 are also expressed in OS tissues, and significantly promote the apoptosis of CD4+ and CD8+ T cells in the TME of OS, and lead to poor prognosis in patients with OS (112,113). Thus, blocking immune checkpoints could further enhance the function of CAR-T cells.
Immunosuppressive cells
Within the TME, there are diverse cell populations that contribute to immune suppression, including myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs) and regulatory T cells (Tregs). Together with tumor cells, these cells orchestrate the control of the tumor and facilitate the production of inhibitory cytokines, chemokines and growth factors (114).
MDSCs are a prominent type of immune-suppressive cells that can hinder the antitumor function of CAR-T cells and can facilitate tumor angiogenesis. Additionally, MDSCs impede T cell proliferation by employing arginase, transforming growth factor (TGF)-β, inducible nitric oxide synthase and IL-10 (115). Moreover, MDSCs can phagocytose antigens, degrade them and present them to CD8+ T cells that are specialized in recognizing such antigens. When MDSCs interact directly with T cells, MDSCs produce a substance called peroxynitrite. This substance modifies certain amino acids in the T-cell receptor and CD8+ molecules on the surface of T cells, which in turn impairs the response of T cells to specific antigen stimulation (115). The activity of MDSCs reduces the effectiveness of the immune response within the TME.
TAMs can be classified into two distinct categories: i) M1 TAMs, which inhibit tumor growth, and ii) M2 TAMs, which promote tumor growth (116). In the majority of cases, tumor cells have the ability to drive the differentiation of TAMs towards the immunosuppressive M2 subtype rather than the immuno-stimulatory M1 subtype. These M2 TAMs play an important role in promoting tumor formation and facilitating the spread of high-grade OS metastasis. This is achieved through the release of immunosuppressive soluble factors such as IL-10, TGF-β2 and C-C motif chemokine ligand (CCL) 22 (117,118).
Tregs play a crucial role in inducing immunosuppression through various mechanisms. Firstly, Tregs express CTLA-4, which inhibits the maturation of antigen-presenting cells by reducing the expression of CD80/86, and hinders the activation of immune responses. Secondly, Tregs consume IL-2, a molecule necessary for the activation of effector T cells, including CD8+ T cells. As Tregs have high-affinity IL-2 receptors but produce minimal IL-2 themselves, this leads to a lack of IL-2 for effector T cells, resulting in their dysfunction. Lastly, Tregs secrete immunosuppressive cytokines like IL-10 and IL-35, directly inhibiting the activation of T cells (119).
Immunosuppressive cytokines
Immunosuppressive cytokines such as IL-10, IL-6 and TGF-β play an essential role in reducing the effectiveness of T cells in fighting against the tumor (120-122). Among them, the most critical inhibitory cytokine is TGF-β, which can act in tumors to stimulate tumor cells to spread further, metastasize and produce cytokines (123). In OS tissue, OS cells can directly release TGF-β1, and improved expression of TGF-β1 is associated with metastasis of high-grade OS (124). Moreover, elevated levels of IL-10 in tumor tissue hinder the production of IL-12 by dendritic cells, which inhibits the cytotoxic T cell response and the activation of NK cells (125). Furthermore, IL-10 hinders the antigen presentation process and suppresses the release of pro-inflammatory cytokines by antigen-presenting cells, which promotes immune tolerance. Elevated levels of IL-10 not only exhibit strong immunosuppressive activity but also contribute to tumor cell proliferation and increased resistance to chemotherapy (126). IL-6 acts not only internally in tumor cells to promote tumor cell proliferation, survival and metastasis, but also externally in other cells present in the TME to promote tumor angiogenesis and evade immune surveillance (127).
Mesenchymal stem cells (MSCs)
MSCs are considered the utmost important factor in promoting OS migration in the TME (128). On the one hand, the interaction between MSCs and tumor cells facilitates angiogenesis and ultimately leads to the formation of new blood vessel networks in tumor tissues (129,130). However, the newly formed blood vessels are often structurally and morphologically abnormal, leading to persistent hypoxia, acidic TME and invasion of tumor cells into blood vessels, which facilitates tumor cell metastasis, and impairs CAR-T cell survival (131). For example, in a rat model of OS, intravenous administration of MSCs had no effect on tumor growth but significantly promoted metastasis to the lung (132). MSCs could stimulate the proliferation of CD4+ T cells and Tregs, while simultaneously causing a decrease in CD8+ T cells (133). Moreover, certain cytokines secreted by MSCs, such as TGF-β, may promote tumor immune escape and debilitate the antitumor immune response (129). Furthermore, MSCs induce the upregulation of indoleamine 2,3-dioxygenase and PD-L1, which are both involved in the immunosuppression of tumors, leading to inhibition of the efficacy of CAR-T cells (133).
Hypoxia and pH
Tissue hypoxia is a characteristic of the OS microenvironment. Hypoxia, or low oxygen conditions, within the TME plays an important role in chemotherapy resistance, tumor advancement and metastasis in OS. Firstly, hypoxia could regulate OS by primarily activating the hypoxia-inducible factor (HIF) (134). HIF is a heterodimer composed of α and β subunits. HIF expression is increased in OS and has the function of regulating the cellular adaptive response to hypoxia by promoting the transcription of various hypoxia-inducible genes. There are three subtypes of HIF-α, namely HIF-1α, HIF-2α and HIF-3α. Among these HIFs, the most important is HIF-1α (135), which promotes the distant metastasis of OS, and the invasion and proliferation of the OS MG63 and U2OS cell lines, which are inhibited when the expression of HIF-1α is reduced (136,137).
Secondly, immune-inflamed 'hot' tumors are characterized by an abundance of CD8+ T cells and the expression of pro-inflammatory cytokines. By contrast, immune desert 'cold' tumors lack CD8+ T cells and instead harbor populations of immunosuppressive cells such as TAMs, MDSCs and Tregs (138). Hypoxia transforms solid tumors into immune deserts by decreasing the ratio of antitumor M1 TAMs to tumor-friendly M2 TAMs (139). Furthermore, hypoxic tumors secrete chemokines such as CXC motif chemokine ligand (CXCL)12, CCL5 and CCL28, which attract immunosuppressive populations such as Tregs and MDSCs (140). Additionally, the oxygen-deprived environment hinders the utilization of glucose by CAR-T cells, impeding their expansion. Otherwise, CAR-T cells would produce abundant lactic acid through glycolysis, and the increased acidity in the TME would inhibit T lymphocyte function, activate MSCs and ultimately promote OS progression (140,141). Other pH-regulatory enzymes induced under hypoxia, such as carbonic anhydrase, Na/H exchanger, solute carrier family 4 member 4 and indoleamine 2,3-dioxygenase, are responsible for TME acidification and tryptophan deficiency, all of which inhibit the activity of T cells (142-144).
Extracellular vesicles
Extracellular vesicles are membrane-bound vesicles that cells release into the surrounding extracellular matrix. They contain various essential signaling molecules, including proteins, nucleic acids, enzymes and other soluble factors. These vesicles play a crucial role in cell communication, angiogenesis and tumor cell proliferation. They achieve this by exchanging information with target cells. Yang et al (145) demonstrated that extracellular vesicles were involved in the progression and spread of OS. Tumor-derived exosomes have been shown in multiple studies to hinder the function of T and NK cells through diverse mechanisms, including induction of T cell apoptosis, enabling OS cells to evade immune surveillance (146). Prudowsky and Yustein (147) demonstrated that extracellular vesicles facilitated the transfer of multidrug resistance 1 mRNA between OS cells, thereby enhancing resistance to Adriamycin.
Metabolism
In the TME, CAR-T cells require sufficient nutrients and energy substances, including glucose, amino acids and glutamine, which are also necessary for tumor cell metabolism, for exerting their antitumor effect. However, tumor cells consume energy more rapidly than T cells, which inhibits T cell proliferation and function due to insufficient nutrient requirements (148).
First, OS cells promote c-Myc expression through platelet-derived growth factor (PDGF)/PDGF receptor β signaling and increase aerobic glycolysis under aerobic conditions (149). Second, TME may also be deprived of essential amino acids. In mouse models of colon and lung cancer, MDSCs with high arginase activity were locally present in tumors and spleens, consuming arginine, and inhibiting T-cell proliferation and cytokine production (150). Arginase-1 is an amino acid-degrading enzyme commonly expressed in the TME, which promotes L-arginine metabolism and inhibits T-lymphocyte responses (151). Third, glutamine is another important factor affecting T-cell function. Since glutamine is required for T cell proliferation, decreased glutamine metabolism in T cells reduces the expression of metabolic regulators and impedes T cell activation (152,153). Thus, it is of utmost importance to maintain a metabolic equilibrium between T and tumor cells, which is vital for the successful management of tumors.
CRS
CRS, a severe adverse effect linked to CAR-T cell therapy, is an inflammatory syndrome triggered by the production of several cytokines by CAR-T cells. This syndrome is considered to be associated with high antitumor activity and tumor burden. The cytokines implicated in CRS consist of IFN-γ, TNF-α, IL-8, IL-10 and macrophage colony-stimulating factor (154,155). The initial indication of CRS typically manifests as fever, which can emerge within hours to days following the infusion of cells. After the initial fever, patients may experience nausea, vomiting, diarrhea, skin rash, delirium, low blood pressure and even severe multi-organ failure (99). A previous study reported serious adverse events, even fatal, caused by CRS (57).
T cell inefficiency
CAR-T cell expansion ability and persistence
Clinical evidence indicates that the expandability and persistence of T cells after infusion are crucial factors for achieving effective cancer therapy (156). However, during CAR-T cell therapy for OS, T cells may become depleted due to various reasons, rendering them unable to fulfill their intended function. A previous study has indicated that a transcription factor known as nuclear receptor subfamily 4 group A number 1 (NR4A1) plays an important role in T cell fatigue. In mouse models with tumor-bearing mice, the injection of NR4A1-CAR-T cells has shown a remarkable ability to impede tumor growth (157). Furthermore, when antigens stimulate naive T cells, they undergo proliferation and differentiate into memory T cells. This process involves several stages, starting with naive T cells and progressing to stem cell memory T cells, central memory T cells, effector memory T cells and finally effector T cells. In mice, it has been observed that the less differentiated subtypes, including naive T cells, stem cell memory T cells and central memory T cells, consistently exhibit higher expansion, longer persistence and stronger antitumor abilities compared with the more differentiated effector memory T cells and finally effector T cells (158). Thus, naive T cells, stem cell memory T cells, central memory T cells, can be employed to augment the antitumor efficacy of CAR-T cells during CAR-T cell production.
CAR-T cell infiltration capability
CAR-T cells targeting solid tumors must penetrate the extracellular matrix and immunosuppressive TME to reach the tumor (159). Some chemokines secreted by solid tumors, including CXCL123, CXCL12 and CXCL5, impede the movement and entry of T cells into tumor sites; however, the lack of appropriate chemokine receptors on T cells hampers their ability to infiltrate the tumor region (160-162). The presence of tumor cells diminishes the quantity and effectiveness of CAR-T cells, significantly compromising their ability to eliminate tumor cells. Hence, enhancing the penetration capacity of CAR-T cells is crucial.
Heterogeneity of OS
In contrast to hematological tumors, OS is a type of solid tumor. The antigens targeted by immunotherapy on solid tumors typically exhibit heterogeneity, varying not only between different types of solid tumors but also between the primary and metastatic stages of the same tumor (163). Thus, careful selection of appropriate TAAs plays a vital role in achieving an effective anti-OS response.
5. Strategies to enhance the efficacy of CAR-T cells
To increase the eliminating efficiency of CAR-T cells and reduce their side effects, a number of measures have been taken based on the shortcomings of CAR-T cells. These measures are mainly aimed at promoting the effect of CAR-T cells, including optimizing CAR-T cell structure, improving the immunological microenvironment, increasing T cell efficiency and preventing CRS.
Optimization of CAR-T cell structure
The utilization of CAR-T cell therapy comes with the inherent risk of off-target effects due to the fact that the TAAs, which CAR-T cells recognize, are not restricted to tumor cells, but can also be detected in normal tissue cells. Additionally, the efficacy of CAR-T cells can be directly reduced by the escape, depletion or loss of tumor antigens. To address these challenges, clinical trials are currently exploring the use of CAR-T cells with two or three target antigens to improve their effectiveness. Therefore, enhancing the anti-OS effect of CAR-T cells could be achieved by optimizing their structure.
Optimization of the antigen recognition domain (ARD)
The main function of the extracellular domain of CAR-T cells is to recognize tumor cell antigens. Researchers have improved the extracellular domain of CAR-T cells to tackle the issues of tumor antigen escape, reduction, loss and off-target effects (164,165). As a result, numerous CAR-T cells have been constructed, including dual-targeted, triple-targeted and tandem CAR-T cells (Fig. 5). Dual- and triple-targeted CAR-T cells refer to the design of two or three separate CARs on a T lymphocyte, where each CAR is specific for different antigens and thus has dual or triple targeting ability. Tandem CAR-T cells are defined as those with two different scFv regions in a single CAR, targeting different antigens.
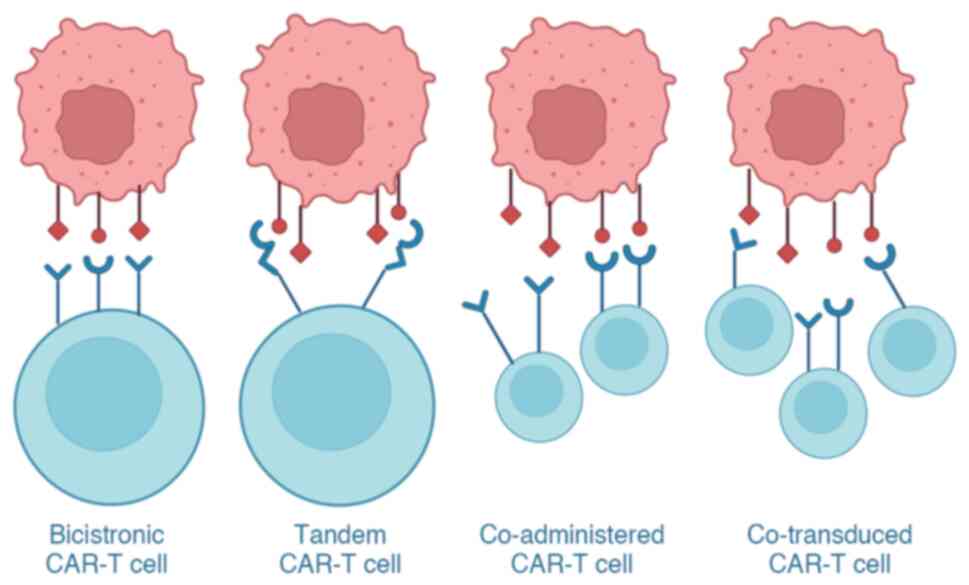
Tian et al (164) demonstrated that, by utilizing dual-target CAR-T cells, which could target two TAAs, it was possible to overcome the heterogeneous expression of these antigens in solid tumors. This approach resulted in improved persistence and enhanced resistance to exhaustion. Moreover, recent research has indicated that the use of innovative tandem CAR-T cells, which target both IL-13Rα2 and EphA2, exhibits significantly higher efficacy in eliminating glioblastoma cells in both laboratory settings and animal models, compared with CAR-T cells that target only one of these antigens (165). To clarify how multi-target CAR-T cells work (for example, whether dual-targeted CAR-T cells are able to eliminate tumors by recognizing only one target antigen, or whether they must recognize both), Boolean logical operations, including 'AND', 'OR' and 'NOT' have been used. CAR-T cells with 'AND' are activated only when both target antigens are recognized, while CAR-T cells with 'OR' can be activated by recognizing either target antigen, and CAR-T cells with 'NOT' avoid eliminating normal cells by stopping their action when a particular antigen is recognized (166). Therefore, the logical gates 'AND', 'OR' and 'NOT' in Boolean logic can improve the recognition and elimination ability of CAR-T cells toward tumor cells and effectively avoid off-target effects. Thus, enhancing the eliminating capacity of CAR-T cells against tumor cells can be achieved by optimizing the ARD, which has been demonstrated to be an effective approach.
Optimization of the HD
The HD or spacer region is located in the extracellular region of CARs. The HD mainly plays the following roles: i) It increases the flexibility of the ARD, which is conducive to target antigen recognition; and ii) both the hinge length and type affect the strength of the activation signal of the CARs and the formation of immune synapses (167,168). To a certain extent, the size of the HD in CAR can be adjusted, reducing the obstacle created by the spatial distance between target and CAR-T cells (169). A long hinge has the advantage of favorable flexibility, which favors the capture of target epitopes close to the membrane with the ARD (170,171). Hudecek et al (172) demonstrated that enhancing the length of the HD could enhance the ability of CAR-T cells to recognize tumors, but a short hinge has the advantage of facilitating the binding of target epitopes far from the membrane (173). The main sources of hinge mentioned in the literature are CD8, CD28, immunoglobulin (Ig)G1, IgG4 and IgD (172,174). Previous studies have revealed that variations in the HD can impact the elimination efficiency, longevity and cytotoxicity of CAR-T cells towards healthy cells (167,175-178). Therefore, optimizing the HD holds great importance for improving the antitumor effect of CAR-T cells.
Optimization of the TMD
The TMD serves to anchor CARs to the cell membrane of T lymphocytes. The TMD is normally formed by CD28, CD8, CD4 and ICOS (179). The TMD has been identified to increase the stability and mobility of CARs (180,181). In first-generation CARs, TMD derived from CD247 promotes T lymphocyte activation (180). The commonly used second- and third-generation CD8- and CD28-derived TMD further improves the stability, mobility and efficacy of CARs compared with the CD247-derived TMD (181-183). Fujiwara et al (184) reported that altering the structure of the TMD affected the expression levels and activity of CARs. The authors also found that modifying the TMD could modulate the function of CAR-T cells without impacting the antigen-binding properties of the ARD or the signal transduction properties of the signal transduction domain (STD). Although previous studies have improved the persistence of T cells, inhibited tumor growth and reduced the incidence of complications such as CRS by optimizing the structure of the TMD of CAR, the role of the TMD needs further investigation (181,182,185). Thus, ongoing optimization of the TMD structure is important for improving the efficacy of CAR-T cells.
Optimization of the intracellular domain
The intracellular domain of CAR-T cells comprises both costimulatory molecules and a STD. Huang et al (186) observed that the costimulation domains significantly enhanced the expansion and long-term persistence of CAR-T cells for therapeutic purposes. The costimulatory domains significantly contributed to the proliferation and persistence of CAR-T cells for therapy. The TNF receptor (CD137, CD27 and OX40) (53,187,188) and CD28 (189,190) families are the most commonly used costimulatory molecules. Exploring the activation mechanism of costimulatory molecules is a crucial approach to identify optimization strategies for CAR-T cells. Currently, CD28 and CD137 are FDA-approved costimulatory molecules for CAR-T cells (191). Although the effectiveness of CAR-T cells with these two costimulatory molecules in treating hematological tumors is similar, there are differences in the eliminating effect, duration and mechanism of CAR-T cells. Specifically, CAR-T cells using CD28 as a costimulatory molecule are characterized by rapid eliminating and high efficacy but low durability (192,193), while CAR-T cells using CD137 as a costimulatory molecule are characterized by moderate and weak eliminating but high durability (193). Several studies have attempted to optimize costimulatory molecules to enhance T lymphocyte activity. Guedan et al (194) identified an amino acid residue in CD28 that promoted T lymphocyte exhaustion. The authors replaced phenylalanine with asparagine in CD28, which delayed T cell exhaustion, increased durability and improved antitumor efficacy. Further studies should combine the advantages and disadvantages of the costimulatory molecules CD28 and CD137, and redesign them to increase antitumor activity. Overall, the signals provided by costimulatory molecules during CAR-T cell activation are critical for T lymphocyte metabolism, survival and efficacy. Therefore, a detailed study of the mechanism of costimulatory molecules would be helpful to construct CAR-T cells with high efficacy, long endurance and low side effects to improve their effect on tumor treatment.
The STD primarily consists of CD247, which is mainly derived from the T cell receptor complex. CD247 contains three immunoreceptor tyrosine-based activation motifs (ITAMs) and is present in almost all CAR structures. Van der Merwe and Dushek (195) reported that the ITAM was an immune receptor activation domain consisting of two Yxx (I/L) and 6-8 amino acids. Gaud et al (196) found that Src family lymphocyte-specific protein tyrosine kinase phosphorylates the tyrosine in the ITAM, which then recruits ZAP70 kinase and further activates signaling molecules such as T lymphocyte linker protein and phospholipase C-γ. In addition, Feucht et al (197) found that altering the second and third ITAMs in CD247 increased the efficacy and durability of CAR-T cells. James (198) noticed that augmenting the number of ITAMs in a second-generation CAR led to an enhancement in the activity of CAR-T cells. Thus, the localization and quantity of ITAMs can influence the functionality of CAR-T cells. A comprehensive investigation of the STD and its associated functions in CAR should therefore help to develop new CAR-T cells and improve their effectiveness against tumors.
To improve the efficacy of tumor cell eradication, various studies have experimented with co-transducing CAR-T cells and employing a combination of two distinct single-targeted CAR-T cell therapies (199-201) (Fig. 5). Co-transduced CAR-T cells are T cells that have been co-transduced with two different lentiviral vectors, with some of the resulting T cells expressing both CARs and some expressing only one CAR (199). Ghorashian et al (200) employed co-transduced CAR-T cells for treating patients with relapsed or refractory ALL, and found that co-transduced CAR-T cells exhibited favorable safety profiles, robust expansion capacity and promising early efficacy. Dual CAR-T cell therapy refers to the administration of two single-targeted CAR-T cells, either simultaneously or sequentially, with each CAR-T cell targeting a distinct antigen (Fig. 5). It has been demonstrated that the sequential infusion of distinct CAR-T cells, each designed to target specific tumor antigens, exhibited promising antitumor effects in clinical trials (201). These findings suggested that the sequential injection of different CAR-T cells may be an effective strategy to combat tumors.
Improvement of the TME
There are numerous ways of improving the TME, and the mechanisms involved are described below (Fig. 4).
Combination with checkpoint inhibitors
Checkpoint inhibitors increase the immune activity of CAR-T cells, reducing immunosuppression caused by increased cytokines and loss of target antigens, leading to activation of inhibited T cells (202). Zheng et al (35) reported that the PD-1 inhibitor nivolumab effectively stopped the spread of OS in mice by boosting the population of CD8+ cells and enhancing the cytotoxic function of CD8+ T cells. Furthermore, PD-1 treatment not only decreased the number of tumor cells and promoted apoptosis in lung metastases of OS, but also made macrophages change from an M2 to an M1 phenotype, resulting in the regression of lung metastases in OS (203). Moreover, Rafiq et al (204) have engineered CAR-T cells capable of producing scFv to block PD-1, and preclinical studies have indicated that these CAR-T cells demonstrate equivalent or improved efficacy compared with combination therapy involving CAR-T cells and PD-1 inhibitors. Kenderian et al (205) reported that immune checkpoint blockers such as TIM-3 combined with CAR T-cell therapy could have synergistic antitumor effects.
Targeting immunosuppressive cells
Immunosuppressive cells present in the TME, such as MDSCs, Tregs and TAMs, hinder the antitumor effectiveness of CAR-T cells.
MDSCs play a critical role as immunosuppressive cells and can be effectively targeted through the following approaches: i) Depletion of MDSCs in the bloodstream and at tumor infiltration sites by using low-dose chemotherapy. Drugs such as 5-fluorouracil, paclitaxel and gemcitabine have been shown to effectively reduce MDSC populations. Combining this low-dose chemotherapy with CAR-T-cell therapy can enhance antitumor responses. By targeting and depleting MDSCs, this combination therapy holds great potential in improving the outcomes of cancer treatment (206-209); ii) preventing the aggregation of MDSCs. A previous study revealed a positive association between MDSC levels and IL-18, indicating that IL-18 promoted the migration of MDSCs into tumor tissue. By administering anti-IL-18 treatment, a significant reduction in MDSCs in both tumors and peripheral blood was observed. This finding suggested that targeting IL-18 could be a favorable approach to improve the effectiveness of immunotherapy in patients with OS (34); iii) counteracting the immunosuppressive function of MDSCs by the use of all-trans-retinoic acid (ATRA). Administering this compound to mice with OS was found to effectively eliminate MDSCs and reduce their inhibitory impact. Additionally, combining ATRA with GD2-CAR-T cell therapy in the management of OS demonstrated significant improvements in antitumor efficacy. This combination approach holds great potential in enhancing the immune response against OS and improving the overall outcome of treatment (74); and iv) promoting the differentiation of MDSCs into a non-suppressive immune state. ATRA was found to have a direct effect on MDSCs, inducing their differentiation into mature myeloid cells such as macrophages and dendritic cells. This process led to a significant decrease in the inhibitory effect of MDSCs on T cells, ultimately promoting an improved immune response (210).
Tregs are another type of immunosuppressive cells. One of the primary approaches in Tregs-based immunotherapy is decreasing the population of Tregs, for example, by applying anti-CD25 monoclonal antibodies to eliminate CD4+ and CD25+ Tregs, and to facilitate the specific eliminating effect of CD8+ T cells in OS. The second approach involves preventing the immunosuppressive effects of Tregs, which express certain proteins that suppress the immune response. These proteins, such as CTLA-4 and PD-L1, can be blocked using specific inhibitors. The third option aims to enhance the activity of effector T cells, which is responsible for suppressing the suppressive effects of Tregs. In immune surveillance, checkpoints inhibit the activation of T cells, leading to the inability of TILs to eliminate cancer cells in OS. Yoshida et al (211) found that checkpoint inhibitors could restore T cell-mediated antitumor responses by releasing the brake and engaging major histocompatibility complex molecules.
TAMs are also immunosuppressive cells in the TME, and targeting TAMs is mainly achieved by switching subtype and reducing the number of TAMs. Blocking the receptor of colony-stimulating factor 1, an important regulator of TAMs recruitment, not only decreases TAMs recruitment to tumors but also enhances the differentiation of TAMs into a proinflammatory M1 phenotype, leading to an increased intratumoral M1/M2 ratio in mice (212). Furthermore, it was revealed that low-dose radiotherapy could reprogram TAMs into M1 TAMs (213,214). Moreover, certain antigens expressed on immunosuppressive cells within the TME could be targeted to eradicate both tumor and immunosuppressive cells, as demonstrated by the specific action of CD123-CAR-T cells against tumor cells and TAMs in Hodgkin lymphoma (215).
Targeting immunosuppressive cytokines
Neutralizing TGF-β, a critical tumor suppressive cytokine, has the potential to amplify the antitumor immune response mediated by CD8+ T cells. Several strategies have been explored based on this concept. Inhibition of TGF-β1 signaling using vactosertib, a TGF-β receptor (TGF-βR)1 inhibitor, has demonstrated a significant reduction in OS cell proliferation both in laboratory settings and in animal models (216). Wallace et al (36) observed that inhibiting TGF-βR led to a significant improvement in the efficacy of adoptive T-cell therapy in animal models of solid tumors. Tang et al (217) demonstrated that removing TGF-βR 2 by using genome editing techniques reduced exhaustion in CAR-T cells and enhanced the effectiveness of mesothelin-CAR-T cells against ovarian cancer cells.
Production of proinflammatory cytokines
Another approach to increase the efficiency of CAR-T cells is to induce them to produce proinflammatory cytokines. Armored CAR-T cells have been hereditarily engineered to produce proinflammatory cytokines, aiming to shield them from the inhibitory effects of the TME. IL-18-secreting CAR-T cells, for example, exhibited increased expansion, persistence and antitumor cytotoxicity compared with CAR-T cells that target only tumor antigens. Additionally, these enhanced CAR-T cells not only altered the TME in mouse lymphomas but also administered IL-18 directly to the tumors. This approach facilitated the recruitment and activation of native antitumor immune effector cells, leading to a robust and comprehensive endogenous antitumor immune response (39). In addition, CAR-T cells that could secrete cytokines such as IL-15 (218) and IL-12 (219) demonstrated amplified proinflammatory capabilities. Thus, CAR-T cells secreting proinflammatory cytokines would be an ideal solution for treating OS.
Regulation of the metabolism
CAR-T cells produce abundant lactate through glycolysis, which increases the acidity of the TME and inhibits T lymphocyte function. Small molecules and metabolites such as cytokines, the glucose analogue 2-deoxy-D-glucose, L-arginine and carnosine have been revealed to affect T-cell metabolism and differentiation. Numerous studies have investigated and confirmed the ability of the cytokines IL-7, IL-15 and IL-21 to limit or interfere with glycolysis (220,221). 2-Deoxy-D-glucose is a glycolytic inhibitor. During in vitro expansion, CD8+ T cells change their differentiation direction in the presence of 2-deoxy-D-glucose to form more memory cells, resulting in long-lasting antitumor function (222). Intracellular L-arginine is significantly reduced after activation of naive T cells, and the additional incorporation of L-arginine into the culture medium causes a shift in metabolism from glycolysis to oxidative phosphorylation. It also increases the level of stem cell memory T cells, resulting in greater antitumor activity (223). In another study, carnosine was found to limit the acidification of extracellular fluid, changing the metabolism of activated T cells from glycolytic to aerobic, ultimately leading to an improved antitumor function of T cells. In addition, transiently elevating the level of carnosine in the culture medium increased lentivirus gene expression (224). Therefore, these results provided ideas to increase the therapeutic efficacy of CAR-T-cell therapy.
Prevention and treatment of CRS
According to the criteria used to evaluate adverse reactions, CRS has been classified into five levels, ranging from mild reactions/grade 1 to mortality/grade 5. The extent of CRS triggered by CAR-T cells is directly associated with the expansion of CAR-T cells and the concentration of IL-6 in the bloodstream (225). Turtle et al (226) conducted a study that revealed that tocilizumab, an anti-human IL-6 receptor monoclonal antibody, was the primary management option for patients experiencing moderate to severe CRS. Li et al (10) emphasized the importance of timely tocilizumab treatment for elderly patients with grade 3 or 2 CRS and comorbidities to prevent severe CRS (grade 4 and 5), with the majority of patients achieving rapid remission post-tocilizumab. Davila et al (227) observed that administering high-dose corticosteroids could effectively impede the proliferation and persistence of CAR-T cells in vivo. As a result, high-dose steroids could also be employed as a management alternative for patients experiencing CRS. However, it is essential to highlight that high-dose steroids are typically reserved for patients with severe and life-threatening CRS who have not responded to tocilizumab treatment.
A previous study constructed inhibitory CARs (iCARs) based on the inhibitory molecules CTLA-4 or PD-1, and showed that these iCARs could block T cell responses activated by their endogenous T cell receptors or activated CARs. This inhibitory effect was transient, and, in the absence of an inhibitory signal, suppressed T cells could be reactivated when exposed to an activating signal. Thus, iCARs may allow T cells to target tumor cells while avoiding attacking normal tissue (37). Moreover, an interesting and feasible approach is the use the tyrosine kinase inhibitor dasatinib, which, in preclinical models, efficiently and temporarily hindered the activation of CAR-T cells. Brief treatment with dasatinib early after infusion of CAR-T cells could prevent lethal CRS in mice (228).
It could therefore be inferred that high-grade CRS may be reversed through early and aggressive treatment. Notably, resolution of CRS could result in an anti-CRS syndrome characterized by hypothermia and bradycardia, accompanied by alterations in cytokine levels (229). Therefore, patients should be monitored for vital signs even after successful CRS treatment.
CAR-T cell targeting of tumor blood vessels
Tumor blood vessels, by being highly perfused, have a crucial role in the progression and metastasis of OS. Thus, CAR-T cells targeting OS blood vessels have attracted the attention of researchers. Previous studies have revealed the existence of pro-angiogenic growth factors in the TME, such as vascular endothelial growth factor (VEGF), with tumor cells exhibiting overexpression of receptors for these factors, which is strongly correlated with adverse prognosis and tumor metastasis (38). Fukumura et al (230) found that VEGFR-2 was overexpressed in some tumor stromal cells. CARs targeting VEGFR-1 and VEGFR-2 have demonstrated promising efficacy in disrupting tumor vasculature and reducing tumor cell proliferation by limiting nutrient and oxygen delivery, and preserving normal tissue (231,232).
Improving the effectiveness of T cells
Promoting the settlement of CAR-T cells
The TME in OS tissue is characterized by a high density of newly formed blood vessels, immunosuppressive cells and inhibitory cytokines. These factors work together to shield tumor cells and hinder the infiltration of CAR-T cells (233). Local drug delivery is considered as one of the most effective measures to tackle this issue, and it can be accomplished by directly administering drugs into the tumor site in various tumor models. Local delivery via intracranial/intravenous drug administration has yielded positive results in models of head and neck squamous cell carcinoma, glioblastoma and colorectal cancer with liver metastases (234,235). Moreover, an alternative method for delivering CAR-T cells into solid tumors involves the use of implantable biopolymer devices. This approach eliminates the need for direct injection into the tumor, allowing for prolonged exposure of tumor cells to high concentrations of immune cells. In mouse models with normal immune function but afflicted with pancreatic cancer and melanoma, the use of a bioactive vector substantially enhanced T cell expansion and function (40).
Runt-related transcription factor 3 (Runx3) protein has been found to promote T cell re-accumulation in tumor tissue. In melanoma mouse models, the expression of the Runx3 gene has been found to effectively promote the accumulation of T cells in tumor tissues and suppress tumor growth (236). In addition, accumulation of collagen and heparan sulfate proteoglycans in tumor tissue hampers the penetration of T cells into the extracellular matrix, but this obstacle can be overcome by overexpressing heparanase. Heparanase, an enzyme naturally present in T cells but absent in CAR-T cells, degrades heparan sulfate proteoglycans. Research conducted by Caruana et al (237) confirmed that CAR-T cells engineered to produce heparanase could essentially reduce tumor volume. In solid tumors, chemokines secreted by the tumor can hinder the penetration and migration of T cells. However, certain chemokine receptors expressed on T cells can recognize these chemokines present in the TME. Therefore, it is possible to enhance the homing and infiltration of CAR-T cells by engineering them to express these specific chemokine receptors. This approach has shown promise in increasing the significance of CAR-T cell therapy (238).
Promoting the persistence of CAR-T cells
A major factor contributing to the inefficacy of CAR-T cell therapy is the notable decline in the persistence of CAR-T cells. This decline is primarily a result of the immune system's impaired ability to identify foreign peptides derived from CAR, which subsequently leads to modifications in T lymphocytes through immune-mediated mechanisms (239,240). Methods to improve the persistence of CAR-T cells mainly include binding to oncolytic viruses and engineering chimeric inverted receptors. Xia et al (41) suggested that combining oncolytic viruses with CAR-T cells improved the activity of CAR-T cells. In addition, it was suggested that being infected with oncolytic viruses could potentially boost the capacity of CAR-T cells to infiltrate tumors, overcome immune system suppression and ultimately enhance their ability to persist (241-243). In addition, engineered chimeric inversion receptors represent a practical way to improve the persistence of CAR-T cells by transforming the immunosuppressive surroundings into an immune-stimulating environment. IL-4 secreted by the tumor exerts an inhibitory impact on T lymphocytes. A novel approach called reverse CAR has been engineered on T lymphocytes, where it incorporates the extracellular domain of IL-4 and the intracellular domain of IL-7. Mohammed et al (105) reported that reverse CAR had the ability to transform the inhibitory signal of T lymphocytes into a stimulatory signal, thereby enhancing the persistence of CAR-T cells. Liu et al (244) engineered a chimeric PD-1 receptor by combining a truncated PD-1 extracellular domain with the transmembrane and internal domains of CD28, transforming the PD-1's inhibitory signal into a stimulating signal to improve the persistence of CAR-T cells. Furthermore, secretion of cytokines by CAR-T cells, such as IL-2, IL-15 and IL-18, has been demonstrated to selectively amplify memory CAR-T cells and enhance their persistence (245). Feucht et al (197) reported that modulating the ITAM of CD247 could enhance CAR signaling, leading to a reduction in T cell exhaustion. Continuous activation signal generated by spontaneous clustering of CAR molecules during in vitro expansion of CAR-T cells resulted in T cell exhaustion, which hampered the therapeutic effectiveness and the persistence of CAR-T cells in vivo. Thus, it would be beneficial to regulate the density of CAR molecules on the cell surface to prevent the aggregation of CARs and subsequent sustained cell activation (246).
6. Combined application of CAR-T cells with chemotherapy or radiotherapy
CAR-T cell therapy for OS continues to face several obstacles. Thus, researchers have made several efforts to boost the effectiveness of CAR-T cell therapy in OS by exploring potential approaches such as integrating it with radiotherapy or chemotherapy (Fig. 6) (18).
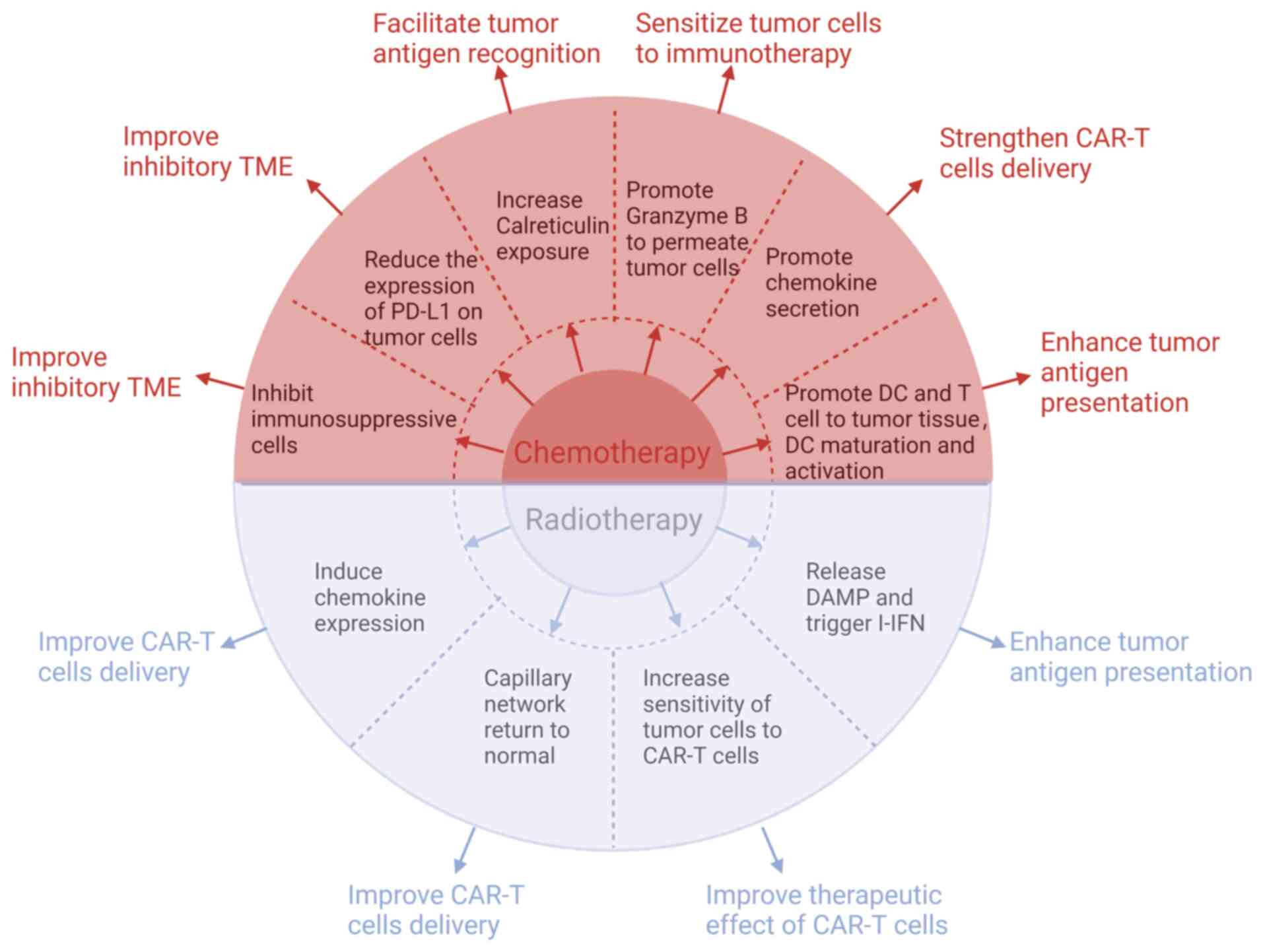
Combined application of CAR-T cells with chemotherapy
The efficacy of local control of CAR-T cells or chemotherapy alone in the treatment of high-grade OS remains suboptimal (247). As a result, certain researchers have proposed that CAR-T cells applied together with chemotherapy could potentially enhance the treatment outcome.
Chemotherapy improves inhibitory TME
Chemotherapeutic agents such as adriamycin, methotrexate, cisplatin and ifosfamide, possess not only tumor-reducing properties but also notable immunomodulatory effects (43). Regular administration of low-dose cyclophosphamide can effectively decrease the quantity and suppressive function of Tregs. Furthermore, it has been observed to augment the presence of CD8+ T cells, which have a pivotal function in triggering immune responses against malignant tumors (248,249). Adriamycin, when combined with CAR-T cells, boosts their antitumor efficacy by suppressing Tregs and MDSCs, leading to a significant inhibition of tumor growth in both mice and humans (250,251). In the context of solid tumors, Seliger and Quandt (252) noted that tumor cells possessed the capability to enhance the levels of immune checkpoint molecules, namely PD-L1 and PD-L2. This mechanism effectively hindered the activity of T and CAR-T cells attempting to penetrate and combat the tumor. Chulanetra et al (43) observed that adriamycin was able to increase the response of OS to GD2-CAR-T cells. This effect was achieved by diminishing the expression of PD-L1, an immune checkpoint, in OS cells. In murine models of pancreatic and prostate cancer, a pretreatment strategy involving cyclophosphamide followed by administration of CAR-T cells led to a significant change in the TME. It effectively shifted the TME from an anti-inflammatory state to a proinflammatory state, thereby stimulating the production of chemokines. These chemokines play a crucial role in attracting CAR-T cells and ultimately revitalize immunologically 'cold' tumors, transforming them into 'hot' tumors that are highly responsive to treatment (253).
Chemotherapy enhances T-cell trafficking to the tumor
Overcoming the obstacle of efficiently transporting T cells to the tumor site remains a critical concern in enhancing the potency of CAR-T cell treatment. Research on mice at preclinical stage indicated that there was a link between limited migration of T cells and diminished levels of chemokine expression (254). CXCL9 and CXCL10 induced by adriamycin could attract NKG2D-CD8+ T cells to OS cells and promote NKG2D-CD8+ T cell homing (250). The study by Srivastava et al (255) revealed that, when mice were pre-treated with oxaliplatin and cyclophosphamide, a notable alteration in the TME was observed. The occurrence triggered an inflammatory reaction, leading to an increased release of CCL5, CXCL9, CXCL10 and CXCL16 by tumor macrophages. This facilitated ROR1-CAR-T cells to enter and effectively target lung tumor cells, leading to a significant improvement in their ability to eliminate them (255). The study by Srivastava et al (255) was not on OS but on lung tumors. However, both lung tumors and OS are solid tumors and both have a similar TME. Hence, further exploration into the combination of chemotherapy and CAR T-cell therapy for enhancing the therapeutic outcomes of OS is a valuable endeavor.
Chemotherapy increases tumor cell sensitivity to immunotherapy
Following targeted chemotherapy, there was a notable increase in the presence of mannose-6-phosphate receptor on cancer cells, which, in turn, allowed granzyme B to enter these cells more effectively. This process not only aided in self-regulation but also heightened the responsiveness of tumor cells to immunotherapy through autophagy (256-258). Parente-Pereira et al (259) conducted a preclinical investigation revealing that combining HER2-CAR-T cells with a low dose of carboplatin not only amplified the sensitivity of tumor cells to HER2-CAR-T cell-induced cytotoxicity but also augmented the overall effectiveness of antitumor treatment. While the mechanism behind increased tumor tissue susceptibility to immunotherapy after chemotherapy remains unclear, Proietti et al (260) have observed improved efficacy in tumor treatment when combining immunotherapy with chemotherapy. Therefore, it could be hypothesized that chemotherapy may improve the ability of CAR T-cell therapy in the treatment of OS and may be a promising direction for future insightful research.
Chemotherapy can enhance tumor antigen recognition and presentation
According to Senovilla et al (261), exposure of calreticulin can be increased by certain chemicals, such as paclitaxel and vinblastine, which leads to enhanced recognition of tumor cells. Additionally, chemotherapeutic agents have the ability to enhance the presentation of tumor antigens. The primary pathways involved are as follows: Firstly, activation of autophagy by certain chemotherapeutic drugs results in the release of ATP in tumor cells. This crucial step subsequently encourages the infiltration of dendritic cells and T lymphocytes into tumors, thus facilitating the presentation of tumor antigens (262). Secondly, chemotherapeutic agents can stimulate dying cancer cells to release damage-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB 1), which could then be recognized by Toll-like receptor 4. This recognition promotes the activation of dendritic cells, leading to a significant increase in the body's ability to fight against tumor cells using T cells (263). Thirdly, chemotherapy prompts tumor or stromal cells to generate type I IFN, which activates dendritic cells, initiating cross priming of T cells and effectively controlling the tumor (264,265). Based on the aforementioned studies, it can be proposed that chemotherapy can increase the antigen presentation of OS cells, which can be more easily recognized and killed by CAR-T cells.
Combined application of CAR-T cells with radiotherapy
Radiotherapy can improve the therapeutic effect of CAR-T cells
Radiotherapy has the dual benefit of not only eliminating tumor cells, but also triggering a targeted immune response against the tumor, offering a potential treatment for both local and distant metastatic tumors (266). DeSelm et al (42) found that administering low-dose total body irradiation can enhance pancreatic adenocarcinoma cells' responsiveness to CAR-T cells, enabling the elimination of tumor cells with a reduced dose of CAR-T cells, consequently reducing CRS incidence and enhancing treatment effectiveness. Thus, it is plausible to consider that radiotherapy may enhance the effect of CAR-T cells in the treatment of OS.
Radiotherapy can enhance tumor antigen presentation
Research conducted by Walle et al (267) highlights how radiotherapy has the ability to amplify tumor antigen presentation, thereby offering a promising avenue for improving cancer therapy. The study conducted by Apetoh et al (268) shed light on the ability of radiotherapy to stimulate apoptosis and necrosis within tumor cells, consequently causing the release of DAMPs. Among these patterns, HMGB 1 was particularly observed. Subsequently, the presence of DAMPs and tumor antigens within the TME can activate type I IFN, thereby initiating a series of innate and adaptive immune responses against tumor cells (269,270). Previous studies have reported that the interplay between innate and adaptive immune responses promotes maturation and activation of dendritic cells to increase tumor antigen presentation (271,272). Thus, radiotherapy may enhance OS tumor antigen presentation, making it more readily recognized and inhibited by CAR-T cells.
Radiotherapy can enhance CAR-T cell infiltration
Effective infiltration of CAR-T cells into the tumor are critical steps in inhibiting OS. Weiss et al (273) demonstrated that the concurrent use of local radiotherapy and NKG2D-CAR-T cells enhanced the trafficking of CAR-T cells to the tumor site, leading to an augmented antitumor response of T cells in glioblastoma. Moreover, previous studies have reported that local radiotherapy can stimulate the production of chemokines such as CXCL9, CXCL10 and CXCL16, which in turn increases the recruitment of T cells into the TME, thereby facilitating tumor-suppressive effects (274,275). In addition, low-dose radiotherapy promoted the secretion of IFN-γ, leading to the normalization of the capillary network, which promoted the infiltration of cytotoxic T lymphocytes into the tumor (276). As a result, radiotherapy appears to have the potential to enhance the trafficking of CAR-T cells to OS, leading to improved treatment outcomes for overall survival.
CAR-T cell therapy plus chemoradiotherapy
Given the crucial role of chemoradiotherapy in treating OS, it represents a novel and innovative approach to amplify the antitumor impact of CAR-T cell therapy through this method. However, CAR-T cell therapy combined with chemoradiotherapy for treating OS still faces numerous problems. Firstly, chemoradiotherapy results in treatment-related toxicities, including effects on the immune system of the host such as lymphopenia (277). Additionally, the utilization of CAR-T cells for the treatment of OS is at an early stage, and the exploration of combining chemoradiotherapy with CAR-T cell therapy for OS requires further investigation. While the mechanism of CAR-T cells in conjunction with chemoradiotherapy for treating OS is not yet fully understood, this approach remains a promising treatment option. In summary, CAR-T cell therapy combined with chemoradiotherapy presents a new and innovative approach to treat OS. However, additional research is required to improve understanding of its clinical efficacy and mechanisms of action.
7. Conclusions
CAR-T cell immunotherapy represents a new alternative strategy for the treatment of OS. A variety of target antigens, including DNAJB8, HER2, CD276, CD166, EphA2, GD2, IL-11Rα, IGF-1R, ROR1, NKG2D, CSPG4 and CD44v6, have been explored for CAR-T cell establishment and OS cell elimination, and have produced encouraging results. However, CAR-T cell therapy in OS still encounters several challenges, including the absence of specific tumor antigens, tumor antigen evasion, inhibitory TME, off-target effects and CRS. Consequently, numerous measures have been undertaken to enhance the effectiveness of CAR-T cells, which encompass structural modifications of CAR-T cells, TME enhancement, improved persistence and homing capabilities, engineering CAR-T cells to target the OS vasculature, and combining CAR-T cells with radiotherapy, chemotherapy or checkpoint inhibitors. In conclusion, CAR-T cell therapy represents a novel method for the treatment of OS, offering an alternative therapeutic option for patients with OS.
Availability of data and materials
Not applicable.
Authors' contributions
TY, YW and YZ wrote parts of the initial draft of this review. WJ modified the initial draft of the manuscript. JJ contributed to the figures. WJ and MW edited the initial draft of this paper and made final changes to the final version before submission. All authors read and approved the final manuscript. Data authentication is not applicable.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
The authors would like to thank Miss Ran Meng (student; Chengde Medical University) and Miss Zhijia Ma (postgraduate; Tianjin Medical University) for identifying relevant literature in the manuscript and for editing the manuscript.
Funding
The present study was supported by the Jilin Provincial Science and Technology Department Project (grant no. YDZJ202301ZYTS032), the Qinhuangdao Science & Technology Bureau (grant no. 201902A119), the Jilin Health Science and Technology Capability Enhancement Project (grant no. 2023JC012) and the Jilin University Bethune Plan Project and Jilin Higher Education Research Project (grant no. JGJX2021D28).
References
|
Misaghi A, Goldin A, Awad M and Kulidjian AA: Osteosarcoma: A comprehensive review. SICOT J. 4:122018. View Article : Google Scholar : PubMed/NCBI | |
|
Dorfman HD and Czerniak B: Bone cancers. Cancer. 75(1 Suppl): S203–S210. 1995. View Article : Google Scholar | |
|
Isakoff MS, Bielack SS, Meltzer P and Gorlick R: Osteosarcoma: Current treatment and a collaborative pathway to success. J Clin Oncol. 33:3029–3035. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Rizzo A, Nannini M, Astolfi A, Indio V, De Iaco P, Perrone AM, De Leo A, Incorvaia L, Di Scioscio V and Pantaleo MA: Impact of chemotherapy in the adjuvant setting of early stage uterine leiomyosarcoma: A systematic review and updated meta-analysis. Cancers (Basel). 12:18992020. View Article : Google Scholar : PubMed/NCBI | |
|
Zhou Y, Yang D, Yang Q, Lv X, Huang W, Zhou Z, Wang Y, Zhang Z, Yuan T, Ding X, et al: Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat Commun. 11:63222020. View Article : Google Scholar : PubMed/NCBI | |
|
Rizzo A, Pantaleo MA, Saponara M and Nannini M: Current status of the adjuvant therapy in uterine sarcoma: A literature review. World J Clin Cases. 7:1753–1763. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Santoni M, Rizzo A, Mollica V, Matrana MR, Rosellini M, Faloppi L, Marchetti A, Battelli N and Massari F: The impact of gender on the efficacy of immune checkpoint inhibitors in cancer patients: The MOUSEION-01 study. Crit Rev Oncol Hematol. 170:1035962022. View Article : Google Scholar : PubMed/NCBI | |
|
Stancovski I, Schindler DG, Waks T, Yarden Y, Sela M and Eshhar Z: Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J Immunol. 151:6577–6582. 1993. View Article : Google Scholar : PubMed/NCBI | |
|
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al: Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 368:1509–1518. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Li W, Wu L, Huang C, Liu R, Li Z, Liu L and Shan B: Challenges and strategies of clinical application of CAR-T therapy in the treatment of tumors-a narrative review. Ann Transl Med. 8:10932020. View Article : Google Scholar : PubMed/NCBI | |
|
Abbas MZ: Strategic use of patent opposition safeguard to improve equitable access to innovative health technologies: A case study of CAR T-cell therapy Kymriah. Glob Public Health. 17:3255–3265. 2022. View Article : Google Scholar | |
|
Astolfi A, Nannini M, Indio V, Schipani A, Rizzo A, Perrone AM, De Iaco P, Pirini MG, De Leo A, Urbini M, et al: Genomic database analysis of uterine leiomyosarcoma mutational profile. Cancers (Basel). 12:21262020. View Article : Google Scholar : PubMed/NCBI | |
|
Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, Stewart AK, Hari P, Htut M, Lesokhin A, et al: Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet. 398:314–324. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Van Oekelen O, Aleman A, Upadhyaya B, Schnakenberg S, Madduri D, Gavane S, Teruya-Feldstein J, Crary JF, Fowkes ME, Stacy CB, et al: Neurocognitive and hypokinetic movement disorder with features of parkinsonism after BCMA-targeting CAR-T cell therapy. Nat Med. 27:2099–2103. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, Liedtke M, Rosenblatt J, Maus MV, Turka A, et al: Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 380:1726–1737. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Garfall AL, Stadtmauer EA, Hwang WT, Lacey SF, Melenhorst JJ, Krevvata M, Carroll MP, Matsui WH, Wang Q, Dhodapkar MV, et al: Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight. 4:e1276842018. View Article : Google Scholar | |
|
Guo B, Chen M, Han Q, Hui F, Dai H, Zhang W, Zhang Y, Wang Y, Zhu H and Han W: CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. J Cell Immunother. 2:28–35. 2016. View Article : Google Scholar | |
|
Zhu J, Simayi N, Wan R and Huang W: CAR T targets and microenvironmental barriers of osteosarcoma. Cytotherapy. 24:567–576. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Boettcher M, Joechner A, Li Z, Yang SF and Schlegel P: Development of CAR T cell therapy in children-A comprehensive overview. J Clin Med. 11:21582022. View Article : Google Scholar : PubMed/NCBI | |
|
Sadelain M, Brentjens R and Rivière I: The basic principles of chimeric antigen receptor design. Cancer Discv. 3:388–398. 2013. View Article : Google Scholar | |
|
Morita R, Nishizawa S, Torigoe T, Takahashi A, Tamura Y, Tsukahara T, Kanaseki T, Sokolovskaya A, Kochin V, Kondo T, et al: Heat shock protein DNAJB8 is a novel target for immunotherapy of colon cancer-initiating cells. Cancer Sci. 105:389–395. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, et al: Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 33:1688–1696. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Picarda E, Ohaegbulam KC and Zang X: Molecular pathways: Targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res. 22:3425–3431. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Wang Y, Yu W, Zhu J, Wang J, Xia K, Liang C and Tao H: Anti-CD166/4-1BB chimeric antigen receptor T cell therapy for the treatment of osteosarcoma. J Exp Clin Cancer Res. 38:1682019. View Article : Google Scholar : PubMed/NCBI | |
|
Hsu K, Middlemiss S, Saletta F, Gottschalk S, McCowage GB and Kramer B: Chimeric antigen receptor-modified T cells targeting EphA2 for the immunotherapy of paediatric bone tumours. Cancer Gene Ther. 28:321–334. 2021. View Article : Google Scholar : | |
|
Reppel L, Tsahouridis O, Akulian J, Davis IJ, Lee H, Fucà G, Weiss J, Dotti G, Pecot CV and Savoldo B: Targeting disialoganglioside GD2 with chimeric antigen receptor-redirected T cells in lung cancer. J Immunother Cancer. 10:e0038972022. View Article : Google Scholar : PubMed/NCBI | |
|
Huang G, Yu L, Cooper LJN, Hollomon M, Huls H and Kleinerman ES: Genetically modified T cells targeting interleukin-11 receptor α-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res. 72:271–281. 2012. View Article : Google Scholar | |
|
Huang X, Park H, Greene J, Pao J, Mulvey E, Zhou SX, Albert CM, Moy F, Sachdev D, Yee D, et al: IGF1R- and ROR1-specific CAR T cells as a potential therapy for high risk sarcomas. PLoS One. 10:e01331522015. View Article : Google Scholar : PubMed/NCBI | |
|
Fernández L, Metais JY, Escudero A, Vela M, Valentín J, Vallcorba I, Leivas A, Torres J, Valeri A, Patiño-García A, et al: Memory T cells expressing an NKG2D-CAR efficiently target osteosarcoma cells. Clin Cancer Res. 23:5824–5835. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Riccardo F, Tarone L, Iussich S, Giacobino D, Arigoni M, Sammartano F, Morello E, Martano M, Gattino F, Maria R, et al: Identification of CSPG4 as a promising target for translational combinatorial approaches in osteosarcoma. Ther Adv Med Oncol. 11:17588359198554912019. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang Y, Ding C, Wang J, Sun G, Cao Y, Xu L, Zhou L and Chen X: Prognostic significance of CD44V6 expression in osteosarcoma: A meta-analysis. J Orthop Surg Res. 10:1872015. View Article : Google Scholar : PubMed/NCBI | |
|
Lin Z, Wu Z and Luo W: Chimeric antigen receptor T-cell therapy: The light of day for osteosarcoma. Cancers (Basel). 13:44692021. View Article : Google Scholar : PubMed/NCBI | |
|
Shah NN and Fry TJ: Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 16:372–385. 2019.PubMed/NCBI | |
|
Guan Y, Zhang R, Peng Z, Dong D, Wei G and Wang Y: Inhibition of IL-18-mediated myeloid derived suppressor cell accumulation enhances anti-PD1 efficacy against osteosarcoma cancer. J Bone Oncol. 9:59–64. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Zheng B, Ren T, Huang Y, Sun K, Wang S, Bao X, Liu K and Guo W: PD-1 axis expression in musculoskeletal tumors and antitumor effect of nivolumab in osteosarcoma model of humanized mouse. J Hematol Oncol. 11:162018. View Article : Google Scholar : PubMed/NCBI | |
|
Wallace A, Kapoor V, Sun J, Mrass P, Weninger W, Heitjan DF, June C, Kaiser LR, Ling LE and Albelda SM: Transforming growth factor-beta receptor blockade augments the effectiveness of adoptive T-cell therapy of established solid cancers. Clin Cancer Res. 14:3966–3974. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Fedorov VD, Themeli M and Sadelain M: PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 5:215ra1722013. View Article : Google Scholar : PubMed/NCBI | |
|
Chen JC, Chang YW, Hong CC, Yu YH and Su JL: The role of the VEGF-C/VEGFRs axis in tumor progression and therapy. Int J Mol Sci. 14:88–107. 2012. View Article : Google Scholar | |
|
Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, Park H, Purdon TJ, Daniyan AF, Spitzer MH and Brentjens RJ: Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 23:2130–2141. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Smith TT, Moffett HF, Stephan SB, Opel CF, Dumigan AG, Jiang X, Pillarisetty VG, Pillai SPS, Wittrup KD and Stephan MT: Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest. 127:2176–2191. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Xia T, Konno H and Barber GN: Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 76:6747–6759. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
DeSelm C, Palomba ML, Yahalom J, Hamieh M, Eyquem J, Rajasekhar VK and Sadelain M: Low-dose radiation conditioning enables CAR T cells to mitigate antigen escape. Mol Ther. 26:2542–2552. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Chulanetra M, Morchang A, Sayour E, Eldjerou L, Milner R, Lagmay J, Cascio M, Stover B, Slayton W, Chaicumpa W, et al: GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am J Cancer Res. 10:674–687. 2020.PubMed/NCBI | |
|
Buka D, Dvořák J, Sitorová V, Hátlová J, Richter I and Sirák I: Changes in the CD8+ density of tumor infiltrating lymphocytes after neoadjuvant radiochemotherapy in patients with rectal adenocarcinom. Klin Onkol. 29:204–209. 2016.In Czech. View Article : Google Scholar | |
|
Makita S, Imaizumi K, Kurosawa S and Tobinai K: Chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma: Opportunities and challenges. Drugs Context. 8:2125672019. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang X, Zhu L, Zhang H, Chen S and Xiao Y: CAR-T cell therapy in hematological malignancies: Current opportunities and challenges. Front Immunol. 13:9271532022. View Article : Google Scholar : PubMed/NCBI | |
|
Kong Y, Tang L, You Y, Li Q and Zhu X: Analysis of causes for poor persistence of CAR-T cell therapy in vivo. Front Immunol. 14:10634542023. View Article : Google Scholar : PubMed/NCBI | |
|
Asmamaw Dejenie T, Tiruneh G/Medhin M, Dessie Terefe G, Tadele Admasu F, Wale Tesega W and Chekol Abebe E: Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum Vaccin Immunother. 18:21142542022. View Article : Google Scholar : PubMed/NCBI | |
|
Farkona S, Diamandis EP and Blasutig IM: Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 14:732016. View Article : Google Scholar : PubMed/NCBI | |
|
García Merino A: Anticuerpos monoclonales. Aspectos básicos. Neurología. 26:301–306. 2011. View Article : Google Scholar | |
|
Lu J, Ding J, Liu Z and Chen T: Retrospective analysis of the preparation and application of immunotherapy in cancer treatment (review). Int J Oncol. 60:122022. View Article : Google Scholar : PubMed/NCBI | |
|
Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, Dishop MK, Kleinerman EE, Pule M, Rooney CM, et al: Immunotherapy for osteosarcoma: Genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther. 17:1779–1787. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Pulè MA, Straathof KC, Dotti G, Heslop HE, Rooney CM and Brenner MK: A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 12:933–941. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Yamada R, Takahashi A, Torigoe T, Morita R, Tamura Y, Tsukahara T, Kanaseki T, Kubo T, Watarai K, Kondo T, et al: Preferential expression of cancer/testis genes in cancer stem-like cells: Proposal of a novel sub-category, cancer/testis/stem gene. Tissue Antigens. 81:428–434. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Nishizawa S, Hirohashi Y, Torigoe T, Takahashi A, Tamura Y, Mori T, Kanaseki T, Kamiguchi K, Asanuma H, Morita R, et al: HSP DNAJB8 controls tumor-initiating ability in renal cancer stem-like cells. Cancer Res. 72:2844–2854. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Watanabe Y, Tsukahara T, Murata K, Hamada S, Kubo T, Kanaseki T, Hirohashi Y, Emori M, Teramoto A, Nakatsugawa M, et al: Development of CAR-T cells specifically targeting cancer stem cell antigen DNAJB8 against solid tumours. Br J Cancer. 128:886–895. 2023. View Article : Google Scholar : | |
|
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM and Rosenberg SA: Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 18:843–851. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Abdou AG, Kandil M, Asaad NY, Dawoud MM, Shahin AA and Abd Eldayem AF: The prognostic role of Ezrin and HER2/neu expression in osteosarcoma. Appl Immunohistochem Mol Morphol. 24:355–363. 2016. View Article : Google Scholar | |
|
Xuan Y, Sheng Y, Zhang D, Zhang K, Zhang Z, Ping Y, Wang S, Shi X, Lian J, Liu K, et al: Targeting CD276 by CAR-T cells induces regression of esophagus squamous cell carcinoma in xenograft mouse models. Transl Oncol. 14:1011382021. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang Y, He L, Sadagopan A, Ma T, Dotti G, Wang Y, Zheng H, Gao X, Wang D, DeLeo AB, et al: Targeting radiation-resistant prostate cancer stem cells by B7-H3 CAR T cells. Mol Cancer Ther. 20:577–588. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Du H, Hirabayashi K, Ahn S, Kren NP, Montgomery SA, Wang X, Tiruthani K, Mirlekar B, Michaud D, Greene K, et al: Antitumor responses in the absence of toxicity in solid tumors by targeting B7-H3 via chimeric antigen receptor T cells. Cancer Cell. 35:221–237.e8. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, Labanieh L, Dhingra S, Leruste A, Majzner RG, et al: Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med. 26:712–719. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Talbot LJ, Chabot A, Funk A, Nguyen P, Wagner J, Ross A, Tillman H, Davidoff A, Gottschalk S and DeRenzo C: A novel orthotopic implantation technique for osteosarcoma produces spontaneous metastases and illustrates dose-dependent efficacy of B7-H3-CAR T cells. Front Immunol. 12:6917412021. View Article : Google Scholar : PubMed/NCBI | |
|
Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, Rietberg SP, Linde MH, Xu P, Rota C, et al: CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res. 25:2560–2574. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Swart GWM: Activated leukocyte cell adhesion molecule (CD166/ALCAM): Developmental and mechanistic aspects of cell clustering and cell migration. Eur J Cell Biol. 81:313–321. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Federman N, Chan J, Nagy JO, Landaw EM, McCabe K, Wu AM, Triche T, Kang H, Liu B, Marks JD and Denny CT: Enhanced growth inhibition of osteosarcoma by cytotoxic polymerized liposomal nanoparticles targeting the alcam cell surface receptor. Sarcoma. 2012:1269062012. View Article : Google Scholar : PubMed/NCBI | |
|
He S, Li S, Guo J, Zeng X, Liang D, Zhu Y, Li Y, Yang D and Zhao X: CD166-specific CAR-T cells potently target colorectal cancer cells. Transl Oncol. 27:1015752023. View Article : Google Scholar | |
|
Kang BH, Jensen KJ, Hatch JA and Janes KA: Simultaneous profiling of 194 distinct receptor transcripts in human cells. Sci Signal. 6:rs132013. View Article : Google Scholar : PubMed/NCBI | |
|
Pasquale EB: Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat Rev Cancer. 10:165–180. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Wykosky J and Debinski W: The EphA2 receptor and ephrinA1 ligand in solid tumors: Function and therapeutic targeting. Mol Cancer Res. 6:1795–1806. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Fritsche-Guenther R, Noske A, Ungethüm U, Kuban RJ, Schlag PM, Tunn PU, Karle J, Krenn V, Dietel M and Sers C: De novo expression of EphA2 in osteosarcoma modulates activation of the mitogenic signalling pathway. Histopathology. 57:836–850. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Doronin II, Vishnyakova PA, Kholodenko IV, Ponomarev ED, Ryazantsev DY, Molotkovskaya IM and Kholodenko RV: Ganglioside GD2 in reception and transduction of cell death signal in tumor cells. BMC Cancer. 14:2952014. View Article : Google Scholar : PubMed/NCBI | |
|
Roth M, Linkowski M, Tarim J, Piperdi S, Sowers R, Geller D, Gill J and Gorlick R: Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer. 120:548–554. 2014. View Article : Google Scholar | |
|
Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, El-Etriby R, Galli S, Tsokos MG, Orentas RJ and Mackall CL: Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res. 4:869–880. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Suri M, Soni N, Okpaleke N, Yadav S, Shah S, Iqbal Z, Alharbi MG, Kalra HS and Hamid P: A deep dive into the newest avenues of immunotherapy for pediatric osteosarcoma: A systematic review. Cureus. 13:e183492021.PubMed/NCBI | |
|
Park JA and Cheung NKV: GD2 or HER2 targeting T cell engaging bispecific antibodies to treat osteosarcoma. J Hematol Oncol. 13:1722020. View Article : Google Scholar : PubMed/NCBI | |
|
Jiang J, Wang R, Yang L, Sha Y, Zhao S, Guo J, Chen D, Zhong Z and Meng F: IL-11Rα-targeted nanostrategy empowers chemotherapy of relapsed and patient-derived osteosarcoma. J Control Release. 350:460–470. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Lokau J, Schoeder V and Garbers C: The role of interleukin-11 in osteosarcoma. Der Pathologe. 41:163–167. 2020.In German. View Article : Google Scholar | |
|
Li YS, Liu Q, He HB and Luo W: The possible role of insulin-like growth factor-1 in osteosarcoma. Curr Probl Cancer. 43:228–235. 2019. View Article : Google Scholar | |
|
Duan Z, Choy E, Harmon D, Yang C, Ryu K, Schwab J, Mankin H and Hornicek FJ: Insulin-like growth factor-I receptor tyrosine kinase inhibitor cyclolignan picropodophyllin inhibits proliferation and induces apoptosis in multidrug resistant osteosarcoma cell lines. Mol Cancer Ther. 8:2122–2130. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Tan X, Fan S, Wu W and Zhang Y: MicroRNA-26a inhibits osteosarcoma cell proliferation by targeting IGF-1. Bone Res. 3:150332015. View Article : Google Scholar : PubMed/NCBI | |
|
Liu Y, Zhu ST, Wang X, Deng J, Li WH, Zhang P and Liu BS: MiR-100 inhibits osteosarcoma cell proliferation, migration, and invasion and enhances chemosensitivity by targeting IGFIR. Technol Cancer Res Treat. 15:NP40–NP48. 2016. View Article : Google Scholar | |
|
Chen G, Fang T, Huang Z, Qi Y, Du S, Di T, Lei Z, Zhang X and Yan W: MicroRNA-133a inhibits osteosarcoma cells proliferation and invasion via targeting IGF-1R. Cell Physiol Biochem. 38:598–608. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Hojjat-Farsangi M, Moshfegh A, Daneshmanesh AH, Khan AS, Mikaelsson E, Osterborg A and Mellstedt H: The receptor tyrosine kinase ROR1-an oncofetal antigen for targeted cancer therapy. Semin Cancer Biol. 29:21–31. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Dai B, Shen Y, Yan T and Zhang A: Wnt5a/ROR1 activates DAAM1 and promotes the migration in osteosarcoma cells. Oncol Rep. 43:601–608. 2020.PubMed/NCBI | |
|
Zhang T, Barber A and Sentman CL: Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res. 66:5927–5933. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Ding H, Yang X and Wei Y: Fusion proteins of NKG2D/NKG2DL in cancer immunotherapy. Int J Mol Sci. 19:1772018. View Article : Google Scholar : PubMed/NCBI | |
|
Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, Schmucker A, Reder J, Sentman CL, Gilham DE, et al: Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res. 7:100–112. 2019. View Article : Google Scholar | |
|
Tao K, He M, Tao F, Xu G, Ye M, Zheng Y and Li Y: Development of NKG2D-based chimeric antigen receptor-T cells for gastric cancer treatment. Cancer Chemother Pharmacol. 82:815–827. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang Y, Li X, Zhang J and Mao L: Novel cellular immunotherapy using NKG2D CAR-T for the treatment of cervical cancer. Biomed Pharmacother. 131:1105622020. View Article : Google Scholar : PubMed/NCBI | |
|
Sun B, Yang D, Dai H, Liu X, Jia R, Cui X, Li W, Cai C, Xu J and Zhao X: Eradication of hepatocellular carcinoma by NKG2D-based CAR-T cells. Cancer Immunol Res. 7:1813–1823. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Wang X, Wang Y, Yu L, Sakakura K, Visus C, Schwab JH, Ferrone CR, Favoino E, Koya Y, Campoli MR, et al: CSPG4 in cancer: multiple roles. Curr Mol Med. 10:419–429. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Rolih V, Barutello G, Iussich S, De Maria R, Quaglino E, Buracco P, Cavallo F and Riccardo F: CSPG4: A prototype oncoantigen for translational immunotherapy studies. J Transl Med. 15:1512017. View Article : Google Scholar : PubMed/NCBI | |
|
Casanova JM, Almeida JS, Reith JD, Sousa LM, Fonseca R, Freitas-Tavares P, Santos-Rosa M and Rodrigues-Santos P: Tumor-infiltrating lymphocytes and cancer markers in osteosarcoma: Influence on patient survival. Cancers (Basel). 13:60752021. View Article : Google Scholar : PubMed/NCBI | |
|
Deng Z, Niu G, Cai L, Wei R and Zhao X: The prognostic significance of CD44V6, CDH11, and β-catenin expression in patients with osteosarcoma. Biomed Res Int. 2013:4961932013. View Article : Google Scholar | |
|
Qiao GL, Song LN, Deng ZF, Chen Y and Ma LJ: Prognostic value of CD44v6 expression in breast cancer: A meta-analysis. Onco Targets Ther. 11:5451–5457. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Saito S, Okabe H, Watanabe M, Ishimoto T, Iwatsuki M, Baba Y, Tanaka Y, Kurashige J, Miyamoto Y and Baba H: CD44v6 expression is related to mesenchymal phenotype and poor prognosis in patients with colorectal cancer. Oncol Rep. 29:1570–1578. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Nakajima K, Taniguchi K and Mutoh KI: Expression of CD44v6 as matrix-associated ectodomain in the bone development. J Vet Med Sci. 72:1017–1022. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, Ye Z and Qian Q: Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci. 15:2548–2560. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Liu B, Yan L and Zhou M: Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am J Cancer Res. 9:228–241. 2019.PubMed/NCBI | |
|
Saifullah MK, Fox DA, Sarkar S, Abidi SM, Endres J, Piktel J, Haqqi TM and Singer NG: Expression and characterization of a novel CD6 ligand in cells derived from joint and epithelial tissues. J Immunol. 173:6125–6133. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Ikeda K and Quertermous T: Molecular isolation and characterization of a soluble isoform of activated leukocyte cell adhesion molecule that modulates endothelial cell function. J Biol Chem. 279:55315–55323. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang Q, Zhang Z, Liu G, Li D, Gu Z, Zhang L, Pan Y, Cui X, Wang L, Liu G, et al: B7-H3 targeted CAR-T cells show highly efficient anti-tumor function against osteosarcoma both in vitro and in vivo. BMC Cancer. 22:11242022. View Article : Google Scholar : PubMed/NCBI | |
|
Majzner RG, Heitzeneder S and Mackall CL: Harnessing the immunotherapy revolution for the treatment of childhood cancers. Cancer Cell. 31:476–485. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, Brenner MK, Fisher WE, Leen AM and Vera JF: Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. 25:249–258. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Anderson KG, Stromnes IM and Greenberg PD: Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell. 31:311–325. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Pardoll DM: The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Wagner LM and Adams VR: Targeting the PD-1 pathway in pediatric solid tumors and brain tumors. Onco Targets Ther. 10:2097–2106. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Hashimoto K, Nishimura S and Akagi M: Characterization of PD-1/PD-L1 immune checkpoint expression in osteosarcoma. Diagnostics (Basel). 10:5282020. View Article : Google Scholar : PubMed/NCBI | |
|
Kawano M, Itonaga I, Iwasaki T and Tsumura H: Enhancement of antitumor immunity by combining anti-cytotoxic T lymphocyte antigen-4 antibodies and cryotreated tumor lysate-pulsed dendritic cells in murine osteosarcoma. Oncol Rep. 29:1001–1006. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Lussier DM, Johnson JL, Hingorani P and Blattman JN: Combination immunotherapy with α-CTLA-4 and α-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J Immunother Cancer. 3:212015. View Article : Google Scholar | |
|
Sun CY, Zhang Z, Tao L, Xu FF, Li HY, Zhang HY and Liu W: T cell exhaustion drives osteosarcoma pathogenesis. Ann Transl Med. 9:14472021. View Article : Google Scholar : PubMed/NCBI | |
|
Liu Y, Luo J, Li Y, Cao J and Wang X: IFNγ and TNFα synergistically promote galectin 9 secretion by human osteosarcoma cells MG-63 to prevent T cell killing. Int J Clin Exp Pathol. 13:2009–2017. 2020. | |
|
Sterner RC and Sterner RM: CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 11:692021. View Article : Google Scholar : PubMed/NCBI | |
|
Gabrilovich DI and Nagaraj S: Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 9:162–174. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Hattinger CM, Salaroglio IC, Fantoni L, Godel M, Casotti C, Kopecka J, Scotlandi K, Ibrahim T, Riganti C and Serra M: Strategies to overcome resistance to immune-based therapies in osteosarcoma. Int J Mol Sci. 24:7992023. View Article : Google Scholar : PubMed/NCBI | |
|
Huang Q, Liang X, Ren T, Huang Y, Zhang H, Yu Y, Chen C, Wang W, Niu J, Lou J and Guo W: The role of tumor-associated macrophages in osteosarcoma progression-therapeutic implications. Cell Oncol (Dordr). 44:525–539. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Dumars C, Ngyuen JM, Gaultier A, Lanel R, Corradini N, Gouin F, Heymann D and Heymann MF: Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma. Oncotarget. 7:78343–78354. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Sugiyama D, Hinohara K and Nishikawa H: Significance of regulatory T cells in cancer immunology and immunotherapy. Exp Dermatol. 32:256–263. 2023. View Article : Google Scholar | |
|
Taylor A, Verhagen J, Blaser K, Akdis M and Akdis CA: Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: The role of T regulatory cells. Immunology. 117:433–442. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Law AMK, Valdes-Mora F and Gallego-Ortega D: Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells. 9:5612020. View Article : Google Scholar : PubMed/NCBI | |
|
Xiao H, Chen L, Luo G, Son H, Prectoni JH and Zheng W: Effect of the cytokine levels in serum on osteosarcoma. Tumor Biol. 35:1023–1028. 2014. View Article : Google Scholar | |
|
Tian B, Du X, Zheng S and Zhang Y: The role of tumor microenvironment in regulating the plasticity of osteosarcoma cells. Int J Mol Sci. 23:161552022. View Article : Google Scholar : PubMed/NCBI | |
|
Lamora A, Talbot J, Bougras G, Amiaud J, Leduc M, Chesneau J, Taurelle J, Stresing V, Le Deley MC, Heymann MF, et al: Overexpression of smad7 blocks primary tumor growth and lung metastasis development in osteosarcoma. Clin Cancer Res. 20:5097–5112. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Shen L, Li J, Liu Q, Song W, Zhang X, Tiruthani K, Hu H, Das M, Goodwin TJ, Liu R and Huang L: Local blockade of interleukin 10 and C-X-C motif chemokine ligand 12 with nano-delivery promotes antitumor response in murine cancers. ACS Nano. 12:9830–9841. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Rossowska J, Anger N, Szczygieł A, Mierzejewska J and Pajtasz-Piasecka E: Reprogramming the murine colon cancer microenvironment using lentivectors encoding shRNA against IL-10 as a component of a potent DC-based chemoimmunotherapy. J Exp Clin Cancer Res. 37:1262018. View Article : Google Scholar : PubMed/NCBI | |
|
Zeng J, Chen S, Li C, Ye Z, Lin B, Liang Y, Wang B, Ma Y, Chai X, Zhang X, et al: Mesenchymal stem/stromal cells-derived IL-6 promotes nasopharyngeal carcinoma growth and resistance to cisplatin via upregulating CD73 expression. J Cancer. 11:2068–2079. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Yang C, Tian Y, Zhao F, Chen Z, Su P, Li Y and Qian A: Bone microenvironment and osteosarcoma metastasis. Int J Mol Sci. 21:69852020. View Article : Google Scholar : PubMed/NCBI | |
|
Chang AI, Schwertschkow AH, Nolta JA and Wu J: Involvement of mesenchymal stem cells in cancer progression and metastases. Curr Cancer Drug Targets. 15:88–98. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Pietrovito L, Leo A, Gori V, Lulli M, Parri M, Becherucci V, Piccini L, Bambi F, Taddei ML and Chiarugi P: Bone marrow-derived mesenchymal stem cells promote invasiveness and transendothelial migration of osteosarcoma cells via a mesenchymal to amoeboid transition. Mol Oncol. 12:659–676. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Lugano R, Ramachandran M and Dimberg A: Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol Life Sci. 77:1745–1770. 2020. View Article : Google Scholar : | |
|
Tsukamoto S, Honoki K, Fujii H, Tohma Y, Kido A, Mori T, Tsujiuchi T and Tanaka Y: Mesenchymal stem cells promote tumor engraftment and metastatic colonization in rat osteosarcoma model. Int J Oncol. 40:163–169. 2012. | |
|
Zhang R, Liu Q, Zhou S, He H, Zhao M and Ma W: Mesenchymal stem cell suppresses the efficacy of CAR-T toward killing lymphoma cells by modulating the microenvironment through stanniocalcin-1. Elife. 12:e829342023. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang L, Song J, Xin X, Sun D, Huang H, Chen Y, Zhang T and Zhang Y: Hypoxia stimulates the migration and invasion of osteosarcoma via up-regulating the NUSAP1 expression. Open Med (Wars). 16:1083–1089. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Lv X, Li J, Zhang C, Hu T, Li S, He S, Yan H, Tan Y, Lei M, Wen M and Zuo J: The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 4:19–24. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Guan G, Zhang Y, Lu Y, Liu L, Shi D, Wen Y, Yang L, Ma Q, Liu T, Zhu X, et al: The HIF-1α/CXCR4 pathway supports hypoxia-induced metastasis of human osteosarcoma cells. Cancer Lett. 357:254–264. 2015. View Article : Google Scholar | |
|
Liu M, Wang D and Li N: MicroRNA-20b downregulates HIF-1α and inhibits the proliferation and invasion of osteosarcoma cells. Oncol Res. 23:257–266. 2016. View Article : Google Scholar | |
|
Galon J and Bruni D: Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 18:197–218. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
de la Cruz-López KG, Castro-Muñoz LJ, Reyes-Hernández DO, García-Carrancá A and Manzo-Merino J: Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front Oncol. 9:11432019. View Article : Google Scholar : PubMed/NCBI | |
|
Kopecka J, Salaroglio IC, Perez-Ruiz E, Sarmento-Ribeiro AB, Saponara S, De Las Rivas J and Riganti C: Hypoxia as a driver of resistance to immunotherapy. Drug Resist Updat. 59:1007872021. View Article : Google Scholar : PubMed/NCBI | |
|
Avnet S, Di Pompo G, Chano T, Errani C, Ibrahim-Hashim A, Gillies RJ, Donati DM and Baldini N: Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via NF-κB activation. Int J Cancer. 140:1331–1345. 2017. View Article : Google Scholar : | |
|
Bobulescu IA, Di Sole F and Moe OW: Na+/H+ exchangers: Physiology and link to hypertension and organ ischemia. Curr Opin Nephrol Hypertens. 14:485–494. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Swietach P, Vaughan-Jones RD and Harris AL: Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 26:299–310. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Chiche J, Brahimi-Horn MC and Pouysségur J: Tumor hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J Cell Mol Med. 14:771–794. 2010. View Article : Google Scholar | |
|
Yang Q, Liu J, Wu B, Wang X, Jiang Y and Zhu D: Role of extracellular vesicles in osteosarcoma. Int J Med Sci. 19:1216–1226. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Chen C, Xie L, Ren T, Huang Y, Xu J and Guo W: Immunotherapy for osteosarcoma: Fundamental mechanism, rationale, and recent breakthroughs. Cancer Lett. 500:1–10. 2021. View Article : Google Scholar | |
|
Prudowsky ZD and Yustein JT: Recent insights into therapy resistance in osteosarcoma. Cancers (Basel). 13:832020. View Article : Google Scholar | |
|
Xie J, Wu H, Dai C, Pan Q, Ding Z, Hu D, Ji B, Luo Y and Hu X: Beyond Warburg effect-dual metabolic nature of cancer cells. Sci Rep. 4:49272014. View Article : Google Scholar | |
|
Tang HY, Guo JQ, Sang BT, Cheng JN and Wu XM: PDGFRβ modulates aerobic glycolysis in osteosarcoma HOS cells via the PI3K/AKT/mTOR/c-Myc pathway. Biochem Cell Biol. 100:75–84. 2022. View Article : Google Scholar | |
|
Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O'Neill A, et al: Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: A mechanism of tumor evasion. Cancer Res. 65:3044–3048. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Rodríguez PC and Ochoa AC: Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol Rev. 222:180–191. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Pietrobon V and Marincola FM: Hypoxia and the phenomenon of immune exclusion. J Transl Med. 19:92021. View Article : Google Scholar : PubMed/NCBI | |
|
Patel CH and Powell JD: Targeting T cell metabolism to regulate T cell activation, differentiation and function in disease. Curr Opin Immunol. 46:82–88. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Schubert ML, Schmitt M, Wang L, Ramos CA, Jordan K, Muller-Tidow C and Dreger P: Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol. 32:34–48. 2021. View Article : Google Scholar | |
|
Almåsbak H, Aarvak T and Vemuri MC: CAR T cell therapy: A game changer in cancer treatment. J Immunol Res. 2016:54746022016. View Article : Google Scholar : PubMed/NCBI | |
|
Maus MV, Grupp SA, Porter DL and June CH: Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 123:2625–2635. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Chen J, López-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP and Rao A: NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 567:530–534. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Klebanoff CA, Gattinoni L and Restifo NP: Sorting through subsets: Which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother. 35:651–660. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang BL, Qin DY, Mo ZM, Li Y, Wei W, Wang YS, Wang W and Wei YQ: Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci China Life Sci. 59:340–348. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, Wang E, Young HA, Murphy PM and Hwu P: Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. 13:1971–1980. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, Fang Z, Zhao K, Konaparthi R, Hua S, et al: Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. 6:80–95. 2016. View Article : Google Scholar : | |
|
Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al: Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 110:20212–20217. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Xia AL, Wang XC, Lu YJ, Lu XJ and Sun B: Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: Challenges and opportunities. Oncotarget. 8:90521–90531. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Tian M, Cheuk AT, Wei JS, Abdelmaksoud A, Chou HC, Milewski D, Kelly MC, Song YK, Dower CM, Li N, et al: An optimized bicistronic chimeric antigen receptor against GPC2 or CD276 overcomes heterogeneous expression in neuroblastoma. J Clin Invest. 132:e1556212022. View Article : Google Scholar : PubMed/NCBI | |
|
Muhammad N, Wang R, Li W, Zhang Z, Chang Y, Hu Y, Zhao J, Zheng X, Mao Q and Xia H: A novel TanCAR targeting IL13Rα2 and EphA2 for enhanced glioblastoma therapy. Mol Ther Oncolytics. 24:729–741. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Han X, Wang Y, Wei J and Han W: Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol. 12:1282019. View Article : Google Scholar : PubMed/NCBI | |
|
Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC and Riddell SR: The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 3:125–135. 2015. View Article : Google Scholar | |
|
Srivastava S and Riddell SR: Engineering CAR-T cells: Design concepts. Trends Immunol. 36:494–502. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Künkele A, Johnson AJ, Rolczynski LS, Chang CA, Hoglund V, Kelly-Spratt KS and Jensen MC: Functional tuning of CARs reveals signaling threshold above which CD8+ CTL antitumor potency is attenuated due to cell fas-FasL-dependent AICD. Cancer Immunol Res. 3:368–379. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, Raubitschek AA, Forman SJ and Press OW: Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 180:7028–7038. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, Arif S, Mather SJ, Taylor-Papadimitriou J, Burchell JM and Maher J: Retargeting of human T cells to tumor-associated MUC1: The evolution of a chimeric antigen receptor. J Immunol. 180:4901–4909. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C and Riddell SR: Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 19:3153–3164. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O'Neill A, Irlam J, Chester KA, Kemshead JT, Shaw DM, et al: The role of extracellular spacer regions in the optimal design of chimeric immune receptors: Evaluation of four different scFvs and antigens. J Immunother. 28:203–211. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, Xue A, Bot A, Scholler N, Mikkilineni L, et al: Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. 26:270–280. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, Yang S and Kochenderfer JN: Function of novel Anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. 25:2452–2465. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Hombach A, Hombach AA and Abken H: Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 Fc 'spacer' domain in the extracellular moiety of chimeric antigen receptors avoids 'off-target' activation and unintended initiation of an innate immune response. Gene Ther. 17:1206–1213. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Almåsbak H, Walseng E, Kristian A, Myhre MR, Suso EM, Munthe LA, Andersen JT, Wang MY, Kvalheim G, Gaudernack G and Kyte JA: Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 22:391–403. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Watanabe N, Bajgain P, Sukumaran S, Ansari S, Heslop HE, Rooney CM, Brenner MK, Leen AM and Vera JF: Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology. 5:e12536562016. View Article : Google Scholar | |
|
Stoiber S, Cadilha BL, Benmebarek MR, Lesch S, Endres S and Kobold S: Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. 8:4722019. View Article : Google Scholar : PubMed/NCBI | |
|
Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M and Gilham DE: The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol. 184:6938–6949. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Guedan S, Posey AD Jr, Shaw C, Wing A, Da T, Patel PR, McGettigan SE, Casado-Medrano V, Kawalekar OU, Uribe-Herranz M, et al: Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. 3:e969762018. View Article : Google Scholar : PubMed/NCBI | |
|
Wan Z, Shao X, Ji X, Dong L, Wei J, Xiong Z, Liu W and Qi H: Transmembrane domain-mediated Lck association underlies bystander and costimulatory ICOS signaling. Cell Mol Immunol. 17:143–152. 2020. View Article : Google Scholar : | |
|
Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, Myklebust JH, Kadapakkam M, Weber EW, Tousley AM, et al: Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 10:702–723. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Fujiwara K, Tsunei A, Kusabuka H, Ogaki E, Tachibana M and Okada N: Hinge and transmembrane domains of chimeric antigen receptor regulate receptor expression and signaling threshold. Cells. 9:11822020. View Article : Google Scholar : PubMed/NCBI | |
|
Yan Z, Cao J, Cheng H, Qiao J, Zhang H, Wang Y, Shi M, Lan J, Fei X, Jin L, et al: A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: A single-arm, phase 2 trial. Lancet Haematol. 6:e521–e529. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, Gao L, Wen Q, Zhong JF, Zhang C and Zhang X: Recent advances in CAR-T cell engineering. J Hematol Oncol. 13:862020. View Article : Google Scholar : PubMed/NCBI | |
|
Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL and Campana D: Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 18:676–684. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Song DG, Ye Q, Poussin M, Harms GM, Figini M and Powell DJ Jr: CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 119:696–706. 2012. View Article : Google Scholar | |
|
Maher J, Brentjens RJ, Gunset G, Riviere I and Sadelain M: Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. 20:70–75. 2002. View Article : Google Scholar | |
|
Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, Lee J, Posey AD Jr, Scholler J, Scholler N, et al: ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 124:1070–1080. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Mullard A: FDA approves first CAR T therapy. Nat Rev Drug Discov. 16:6692017. | |
|
Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, et al: Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 106:3360–3365. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, Plotkin J and Sadelain M: Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 28:415–428. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Guedan S, Madar A, Casado-Medrano V, Shaw C, Wing A, Liu F, Young RM, June CH and Posey AD Jr: Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J Clin Invest. 130:3087–3097. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
van der Merwe PA and Dushek O: Mechanisms for T cell receptor triggering. Nat Rev Immunol. 11:47–55. 2011. View Article : Google Scholar | |
|
Gaud G, Lesourne R and Love PE: Regulatory mechanisms in T cell receptor signalling. Nat Rev Immunol. 18:485–497. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, Dobrin A, Cabriolu A, Hamieh M and Sadelain M: Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 25:82–88. 2019. View Article : Google Scholar : | |
|
James JR: Tuning ITAM multiplicity on T cell receptors can control potency and selectivity to ligand density. Sci Signal. 11:eaan10882018. View Article : Google Scholar : PubMed/NCBI | |
|
Bachiller M, Dobaño-López C, Rodríguez-García A, Castellsagué J, Gimenez-Alejandre M, Antoñana-Vildosola A, Martin-Antonio B, Delgado J, Pérez-Galán P, Juan M, et al: Co-Transduced CD19/BCMA dual-targeting CAR-T cells for the treatment of non-hodgkin lymphoma. Blood. 140(Suppl 1): S7386–S7387. 2022. View Article : Google Scholar | |
|
Ghorashian S, Lucchini G, Richardson R, Nguyen K, Terris C, Oporto-Espuelas M, Yeung J, Pinner D, Chu J, Williams L, et al: Dual antigen targeting with co-transduced CD19/22 CAR T cells may prevent antigen-negative relapse after CAR T cell therapy for relapsed/refractory ALL. Blood. 140(Suppl 1): S10352–S10354. 2022. View Article : Google Scholar | |
|
Wang L, Tan Su Yin E, Zhao H, Ni F, Hu Y and Huang H: CAR-T cells: The Chinese experience. Expert Opin Biol Ther. 20:1293–1308. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Sun C, Dotti G and Savoldo B: Utilizing cell-based therapeutics to overcome immune evasion in hematologic malignancies. Blood. 127:3350–3359. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Dhupkar P, Gordon N, Stewart J and Kleinerman ES: Anti-PD-1 therapy redirects macrophages from an M2 to an M1 phenotype inducing regression of OS lung metastases. Cancer Med. 7:2654–2664. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, Song M, Miele MM, Li Z, Wang P, et al: Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 36:847–856. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Kenderian SS, Ruella M, Shestova O, Klichinsky M, Kim M, Porter DL, June CH and Gill S: Identification of PD1 and TIM3 As checkpoints that limit chimeric antigen receptor T cell efficacy in leukemia. Biol Blood Marrow Transplant. 22(3 Suppl): S19–S21. 2016. View Article : Google Scholar | |
|
Suzuki E, Kapoor V, Jassar AS, Kaiser LR and Albelda SM: Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 11:6713–6721. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Sevko A, Michels T, Vrohlings M, Umansky L, Beckhove P, Kato M, Shurin GV, Shurin MR and Umansky V: Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma mode. J Immunol. 190:2464–2471. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Eriksson E, Wenthe J, Irenaeus S, Loskog A and Ullenhag G: Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med. 14:2822016. View Article : Google Scholar | |
|
Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rébé C and Ghiringhelli F: 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 70:3052–3061. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Kusmartsev S, Su Z, Heiser A, Dannull J, Eruslanov E, Kübler H, Yancey D, Dahm P and Vieweg J: Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res. 14:8270–8278. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Yoshida K, Okamoto M, Sasaki J, Kuroda C, Ishida H, Ueda K, Ideta H, Kamanaka T, Sobajima A, Takizawa T, et al: Anti-PD-1 antibody decreases tumour-infiltrating regulatory T cells. BMC Cancer. 20:252020. View Article : Google Scholar : PubMed/NCBI | |
|
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al: CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 19:1264–1272. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L, et al: Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 24:589–602. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Nadella V, Singh S, Jain A, Jain M, Vasquez KM, Sharma A, Tanwar P, Rath GK and Prakash H: Low dose radiation primed iNOS + M1macrophages modulate angiogenic programming of tumor derived endothelium. Mol Carcinog. 57:1664–1671. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, Kraft DO, Feldman M, Wasik MA, June CH and Gill S: Overcoming the immunosuppressive tumor microenvironment of Hodgkin lymphoma using chimeric antigen receptor T cells. Cancer Discov. 7:1154–1167. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Choi SH, Myers J, Tomchuck S, Bonner M, Eid S, Kingsley D, VanHeyst K, Kim SJ, Kim BG and Huang AY: Oral TGF-βR1 inhibitor vactosertib promotes osteosarcoma regression by targeting tumor proliferation and enhancing anti-tumor immunity. Res Sq. rs.3.rs-27092822023. | |
|
Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, Wei XF, Han W and Wang H: TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 5:e1339772020. View Article : Google Scholar | |
|
Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, Balyasnikova IV and Gottschalk S: Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. 5:571–581. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Chmielewski M, Kopecky C, Hombach AA and Abken H: IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 71:5697–5706. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Loschinski R, Böttcher M, Stoll A, Bruns H, Mackensen A and Mougiakakos D: IL-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget. 9:13125–13138. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Huang Y, Si X, Shao M, Teng X, Xiao G and Huang H: Rewiring mitochondrial metabolism to counteract exhaustion of CAR-T cells. J Hematol Oncol. 15:382022. View Article : Google Scholar : PubMed/NCBI | |
|
Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al: Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 123:4479–4488. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, et al: L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 167:829–842.e13. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Ghassemi S, Martinez-Becerra FJ, Master AM, Richman SA, Heo D, Leferovich J, Tu Y, García-Cañaveras JC, Ayari A, Lu Y, et al: Enhancing chimeric antigen receptor T cell anti-tumor function through advanced media design. Mol Ther Methods Clin Dev. 18:595–606. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al: T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet. 385:517–528. 2015. View Article : Google Scholar | |
|
Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, Hawkins R, Chaney C, Cherian S, Chen X, et al: Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 8:355ra1162016. View Article : Google Scholar : PubMed/NCBI | |
|
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al: Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 6:224ra252014. View Article : Google Scholar : PubMed/NCBI | |
|
Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, Mades A, Sadelain M, Einsele H and Hudecek M: The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. 11:eaau59072019. View Article : Google Scholar : PubMed/NCBI | |
|
Varadarajan I and Lee DW: Management of T-cell engaging immunotherapy complications. Cancer J. 25:223–230. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Fukumura D, Xavier R, Sugiura T, Chen Y, Park EC, Lu N, Selig M, Nielsen G, Taksir T, Jain RK and Seed B: Tumor induction of VEGF promoter activity in stromal cells. Cell. 94:715–725. 1998. View Article : Google Scholar : PubMed/NCBI | |
|
Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA, Feldman SA, Restifo NP and Rosenberg SA: Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest. 120:3953–3968. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Wang W, Ma Y, Li J, Shi HS, Wang LQ, Guo FC, Zhang J, Li D, Mo BH, Wen F, et al: Specificity redirection by CAR with human VEGFR-1 affinity endows T lymphocytes with tumor-killing ability and anti-angiogenic potency. Gene Ther. 20:970–978. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Slaney CY, Kershaw MH and Darcy PK: Trafficking of T cells into tumors. Cancer Res. 74:7168–7174. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
van Schalkwyk MC, Papa SE, Jeannon JP, Guerrero Urbano T, Spicer JF and Maher J: Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurre head and neck cancer. Hum Gene Ther Clin Dev. 24:134–142. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Sridhar P and Petrocca F: Regional delivery of chimeric antigen receptor (CAR) T-cells for cancer therapy. Cancers (Basel). 9:922017. View Article : Google Scholar : PubMed/NCBI | |
|
Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, et al: Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumors. Nature. 552:253–257. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, Ittmann MM, Marchetti D and Dotti G: Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 21:524–529. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM and Foster AE: Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 33:780–788. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, Zhao Y, Kalos M and June CH: T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 1:26–31. 2013. View Article : Google Scholar | |
|
Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ, et al: Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 112:2261–2271. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Ajina A and Maher J: Prospects for combined use of oncolytic viruses and CAR T-cells. Review. J Immunother Cancer. 5:902017. View Article : Google Scholar | |
|
Kim DS, Dastidar H, Zhang C, Zemp FJ, Lau K, Ernst M, Rakic A, Sikdar S, Rajwani J, Naumenko V, et al: Smac mimetics and oncolytic viruses synergize in driving anticancer T-cell responses through complementary mechanisms. Nat Commun. 8:3442017. View Article : Google Scholar : PubMed/NCBI | |
|
Scott EM, Duffy MR, Freedman JD, Fisher KD and Seymour LW: Solid tumor immunotherapy with T cell engager-armed oncolytic viruses. Macromol Biosci. 18:17001872018. View Article : Google Scholar | |
|
Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y and Moon EK: A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 76:1578–1590. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Chmielewski M and Abken H: TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv Cell Gene Ther. 3:e842020. View Article : Google Scholar | |
|
Zhang H, Zhao P and Huang H: Engineering better chimeric antigen receptor T cells. Exp Hematol Oncol. 9:342020. View Article : Google Scholar : PubMed/NCBI | |
|
Köksal H, Müller E, Inderberg EM, Bruland Ø and Wälchli S: Treating osteosarcoma with CAR T cells. Scand J Immunol. 89:e127412019. View Article : Google Scholar | |
|
Noordam L, Kaijen MEH, Bezemer K, Cornelissen R, Maat LAPWM, Hoogsteden HC, Aerts JGJV, Hendriks RW, Hegmans JPJJ and Vroman H: Low-dose cyclophosphamide depletes circulating naïve and activated regulatory T cells in malignant pleural mesothelioma patients synergistically treated with dendritic cell-based immunotherapy. Oncoimmunology. 7:e14743182018. View Article : Google Scholar | |
|
Ge Y, Domschke C, Stoiber N, Schott S, Heil J, Rom J, Blumenstein M, Thum J, Sohn C, Schneeweiss A, et al: Metronomic cyclophosphamide treatment in metastasized breast cancer patients: Immunological effects and clinical outcome. Cancer Immunol Immunother. 61:353–362. 2012. View Article : Google Scholar | |
|
Hu J, Sun C, Bernatchez C, Xia X, Hwu P, Dotti G and Li S: T-cell homing therapy for reducing regulatory T cells and preserving effector T-cell function in large solid tumors. Clin Cancer Res. 24:2920–2934. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, Katsanis E and Larmonier N: Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 74:104–118. 2014. View Article : Google Scholar | |
|
Seliger B and Quandt D: The expression, function, and clinical relevance of B7 family members in cancer. Cancer Immunol Immunother. 61:1327–1341. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Murad JP, Tilakawardane D, Park AK, Lopez LS, Young CA, Gibson J, Yamaguchi Y, Lee HJ, Kennewick KT, Gittins BJ, et al: Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol Ther. 29:2335–2349. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Kohli K, Pillarisetty VG and Kim TS: Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 29:10–21. 2022. View Article : Google Scholar : | |
|
Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, Garrison SM, Specht JM, Lee SM, Amezquita RA, et al: Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves antitumor efficacy when combined with checkpoint blockade. Cancer Cell. 39:193–208.e10. 2021. View Article : Google Scholar | |
|
Motyka B, Korbutt G, Pinkoski MJ, Heibein JA, Caputo A, Hobman M, Barry M, Shostak I, Sawchuk T, Holmes CF, et al: Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell. 103:491–500. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Ramakrishnan R, Huang C, Cho HI, Lloyd M, Johnson J, Ren X, Altiok S, Sullivan D, Weber J, Celis E and Gabrilovich DI: Autophagy induced by conventional chemotherapy mediates tumor cell sensitivity to immunotherapy. Cancer Res. 72:5483–5493. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Trapani JA, Sutton VR, Thia KYT, Li YQ, Froelich CJ, Jans DA, Sandrin MS and Browne KA: A clathrin/dynaminand mannose-6-phosphate receptor-independent pathway for granzyme B-induced cell death. J Cell Biol. 160:223–233. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Parente-Pereira AC, Whilding LM, Brewig N, van der Stegen SJ, Davies DM, Wilkie S, van Schalkwyk MC, Ghaem-Maghami S and Maher J: Synergistic chemoimmunotherapy of epithelial ovarian cancer using ErbB-Retargeted T cells combined with carboplatin. J Immunol. 191:2437–2445. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Proietti E, Moschella F, Capone I and Belardelli F: Exploitation of the propulsive force of chemotherapy for improving the response to cancer immunotherapy. Mol Oncol. 6:1–14. 2012. View Article : Google Scholar | |
|
Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M, et al: An immunosurveillance mechanism controls cancer cell ploidy. Science. 337:1678–1684. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Séror C, Métivier D, Perfettini JL, Zitvogel L and Kroemer G: Chemotherapy induces ATP release from tumor cells. Cell Cycle. 8:3723–3728. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, Breckpot K, Brough D, Chaurio R, Cirone M, et al: Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. 6:5882015. View Article : Google Scholar : PubMed/NCBI | |
|
Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al: Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 20:1301–1309. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Vierboom MP, Bos GM, Ooms M, Offringa R and Melief CJ: Cyclophosphamide enhances anti-tumor effect of wild-type p53-specific CTL. Int J Cancer. 87:253–260. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Higgins JP, Bernstein MB and Hodge JW: Enhancing immune responses to tumor-associated antigens. Cancer Biol Ther. 8:1440–1449. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Walle T, Martinez Monge R, Cerwenka A, Ajona D, Melero I and Lecanda F: Radiation effects on antitumor immune responses: Current perspectives and challenges. Ther Adv Med Oncol. 10:17588340177425752018. View Article : Google Scholar : PubMed/NCBI | |
|
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al: Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 13:1050–1059. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Crouse J, Kalinke U and Oxenius A: Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol. 15:231–242. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu YX and Auh SL: The efficacy of radiotherapy relies upon induction of type I interferon-dependent innate and adaptive immunity. Cancer Res. 71:2488–2496. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al: Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 208:1989–2003. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM and Gajewski TF: Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 208:2005–2016. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Weiss T, Weller M, Guckenberger M, Sentman CL and Roth P: NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res. 78:1031–1043. 2018. View Article : Google Scholar | |
|
Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, Babb JS, Schneider RJ, Formenti SC, Dustin ML and Demaria S: Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 181:3099–3107. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, Yu P, Fu YX, Weichselbaum RR, Rowley DA, et al: Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 204:49–55. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Ganss R, Ryschich E, Klar E, Arnold B and Hämmerling GJ: Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 62:1462–1470. 2002.PubMed/NCBI | |
|
Yovino S and Grossman SA: Severity, etiology and possible consequences of treatment-related lymphopenia in patients with newly diagnosed high-grade gliomas. CNS Oncol. 1:149–154. 2012. View Article : Google Scholar |