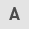SENP1 attenuates hypoxia‑reoxygenation injury in liver sinusoid endothelial cells by relying on the HIF‑1α signaling pathway
- Authors:
- Published online on: February 28, 2024 https://doi.org/10.3892/mmr.2024.13188
- Article Number: 64
-
Copyright: © Qing et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
A number of pathological injuries, such as infection and ischemia-reperfusion injury (I/R), are common in liver transplantation and partial hepatectomy (1–3). Furthermore, I/R is a common pathophysiological process (1). Liver parenchymal and non-parenchymal cells undergo apoptosis or are lysed under ischemic and hypoxic conditions, profoundly affecting the recovery of postoperative liver function (1–3). Liver failure may be the outcome of such severe damage, subsequently endangering the life of the patient (1–4). The mechanisms underlying early I/R injury include Küpffer cell activation, hepatocyte swelling and hepatic microcirculation dysfunction, and liver sinusoidal endothelial cells (LSECs) have an important role in regulating hepatic microcirculation (5–11). Extensive apoptosis and a loss of normal function due to I/R injury may lead to further injury, apoptosis and disrupted liver regeneration (5–7). Therefore, studying the specific mechanism underlying hypoxia-reoxygenation (H-R) injury in LSECs is important to reduce the incidence and severity of hepatic I/R injury.
LSECs have a unique shape, including a very thin cytoplasmic extension and a perforated membrane, termed fenestration. Cells with normal fenestration are considered to be in the differentiation state (12–14). By contrast, a reduction in or abrogation of fenestration is termed capillarization, and LSECs in the dedifferentiation state are associated with hepatic stellate cell activation, liver fibrosis, liver ischemia and hypoxia-effect aggravation (12–14). LSECs with healthy fenestrae play a normal role, and therefore, it is important to maintain the fenestration of LSECs to reduce liver I/R injury and promote liver regeneration. The maintenance of LSEC fenestration requires the participation of both paracrine and autocrine cell signals (13). Vascular endothelial growth factor (VEGF), which has a key role in fenestration regulation, is mediated through nitric oxide (NO)-dependent and NO-independent pathways (15,16). VEGF may also prevent the capillarization of LSECs (16).
In our previous study, it was demonstrated that LESC proliferation and fenestration maintenance is mediated by regulation of the Sentrin/SUMO-specific protease 1 (SENP1)/hypoxia-inducible factor-1α (HIF-1α)/VEGF signaling axis under hypoxic conditions (17). The ubiquitin-specific proteases, SENPs, have an important function in the posttranslational SUMO modification of proteins (17,18). Furthermore, SENP1 is highly sensitive to hypoxia and can regulate the stability of HIF-1α in the nucleus by desumoylation (17–19). In addition, SENP1 and HIF-1α can promote the actions of each other, forming a positive feedback loop and, through this feedback loop, HIF-1α activates the expression of VEGF (17–19). However, this adaptation of cells to hypoxia has only been identified in the hepatic sinusoid (19), and whether this mechanism underlies the effect mediated by LSECs in H-R injury to reduce damage is unknown.
In the present study, it was found that LSECs upregulated the expression of SENP1 and HIF-1α following H-R. However, whether these proteins have a role in the H-R injury of LSECs has not, to the best of our knowledge, been studied. Therefore, the present study utilized mouse LSECs to construct an in vitro H-R model to explore the relationship between SENP1 and LSEC H-R injury, and to explore the specific mechanism of action to provide new targets for the clinical reduction of hepatic I/R injury.
Materials and methods
Cell culture and establishment of an LSEC H-R model
Mouse primary LSECs were purchased from iCell Technologies, Inc. This company obtained LSECs from male C57BL/6 mice by digesting dissected liver tissue with elastase and collagenase and then culturing the cells at 37°C with 5% CO2 in primary endothelial cell culture medium (iCell Bioscience, Inc.). In the present study, LSECs were inoculated in a 6-well culture plate and the culture medium was changed every 24 h. When the cells reached 90% confluency in each well, H-R was performed in a closed chamber at 37°C. For this, the cells were cultured under anoxic conditions [a N2/CO2 (95:5) gas mixture] for 6 h and were then reoxygenated in an air/CO2 (95:5) gas mixture for 24 h. The cells were maintained in low serum (<5%) culture medium (iCell Bioscience, Inc.). The cells in the control group were exposed to normoxia [air/CO2 (95/5)] (20).
Reagents
Rabbit antibodies against SENP1 (cat. no. AF0275) and HIF-1α (cat. no. AF1009) were purchased from Affinity Biosciences. Rabbit antibodies against heme oxygenase (HO-1; cat. no. ab189491), Bax (cat. no. ab32503), Bcl-2 (cat. no. ab182858), cleaved-caspase-3 (cat. no. ab2302), β-actin (cat. no. ab8227) and GAPDH (cat. no. ab9485) were purchased from Abcam. Goat anti-rabbit HRP-conjugated secondary antibodies were purchased from Proteintech Group, Inc. (cat. no. PR30012). Dimethyloxalylglycine (DMOG) was purchased from MedChemExpress. Short interfering (si)-HIF-1α, si-SENP1 and control oligonucleotides were synthesized by Shanghai GenePharma Co., Ltd. These siRNAs were transfected into LSECs by Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.).
Experimental grouping and study process
First, LSECs were randomly divided into two groups for an in vitro study: A normoxic group (group A) and an H-R group (group B). The cells in group B were further subdivided into a control group, which was an H-R injury only group; a negative control (NC) group, in which H-R cells were transfected with si-NC; an si-SENP1 group, in which H-R cells were transfected with si-SENP1; and an si-SENP1 + HIF-1α agonist rescue group, in which H-R cells were transfected with si-SENP1 and then treated with an HIF-1α agonist (DMOG). After 24 h of culture, the cells in each group were analyzed using a scanning electron microscope to determine the effect of H-R on LSEC fenestration. The apoptosis and proliferation rates of the LSECs in each group were measured by flow cytometry and Cell Counting Kit-8 (CCK-8) assay, respectively, to study the effect of H-R on these processes. Furthermore, with an identical procedure, the expression levels of SENP1, HIF-1α, VEGF, HO-1, Bax, Bcl-2 and Caspase-3 in the normoxic and H-R groups were measured.
Scanning electron microscopy examination
After LSECs were seeded onto glass slides, the cells were treated according to the experimental protocols, quickly fixed with 3% glutaraldehyde at 4°C for 2 h and then stored at 4°C overnight. Then, the cells were fixed in a 1% osmium tetroxide (pH 7.4) acetate buffer solution at 4°C for 1 h, dehydrated with a series of ethanol solutions, dried in a critical-point device and then plated onto gold in a vacuum coating device. The fenestration of the cells in each experimental group was determined by observation with a scanning electron microscope (SU8010; Hitachi, Ltd.) with a 1.5 kV acceleration voltage and at ×60,000 magnification.
Cell viability determination by CCK-8 assay
The CCK-8 cell viability assay kit contains WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2rec-4-disulfophenyl)-2H-tetrazolium-monosodium salt; CAS:193149-74-5]. In the presence of an electronic carrier, WST-8 is reduced by intracellular dehydrogenase to form a water-soluble orange methylene dye that can be dissolved in tissue culture medium. The amount of dye produced is proportional to the number of living cells. Therefore, CCK-8 method is a highly sensitive and non-radioactive colorimetric method for determining the number of living cells in cell proliferation experiments. An LSEC suspension (100 µl of 5×103 cells/well) was inoculated into a 96-well plate. When the confluency reached 70%, the cells were cultured in DMEM/F12 without fetal bovine serum (FBS) for 24 h. Then, the culture plate was incubated in an anoxic or normoxic incubator and the medium was replaced with DMEM containing FBS (iCell Bioscience, Inc.). The culture plates were incubated (5 replicate wells for each time point) for the appropriate time (specifically grouped according to the experimental protocol). Then, 10 µl CCK-8 solution (from a CCK-8 detection kit; Peptide Institute, Inc.) was added to the medium and incubated for 2 h. The absorbance was then measured at 450 nm using an automatic microplate reader (EL309; BioTek; Agilent Technologies, Inc.). All experiments were repeated at least three times.
Detection of apoptosis by flow cytometry
Apoptosis was analyzed using an Annexin V-FITC Apoptosis Detection Kit (BD Biosciences). After treatment, the cells were harvested and precipitated from the solution according to the aforementioned experimental methods. Binding buffer (100 µl) was added to each cell sample, and the samples were transferred to a flow tube. Then, 10 µl propidium iodide and 5 µl Annexin V/FITC solution was added to the cells and incubated at room temperature for 15 min. Finally, 900 µl 0.01 M PBS (1X PBS) was added to each cell sample before flow cytometry analysis. Apoptosis was measured using a FACScan flow cytometer (BD Biosciences) and analyzed using FlowJo software (version 10.0; FlowJo LLC). Apoptosis portion (%)=Q2 (late stage apoptotic cells) + Q3 (early apoptotic cells).
si-SENP1 transfection into LSECs
First, the cells were inoculated into culture plates, and the target sequence (sense: 5′-GCAGUUCUGUGUAGCGAAATT-3′, antisense: 5′-UUUCGCUACACAGAACUGCTT-3′) with the highest transfection efficiency and the appropriate transfection conditions (mass, 10 pmol; 37°C for 48 h) were established based on the manufacturer's instructions. The negative control sequence was 5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-ACGUGACACGUUCGGAGAATT-3′. After which, the transfected cells were divided into groups according to the aforementioned experimental grouping, and stably transfected cells were used in the primary and follow-up experiments. The time interval between transfection and subsequent experimentation was 12–24 h.
Western blotting (WB)
The cells in each group were cultured under the aforementioned normoxic or H-R conditions and then collected for WB analysis. First, the proteins were extracted by radioimmunoprecipitation assay RIPA lysis buffer (cat. no. P0013B) and 1% PMSF (cat. no. ST506; both Beyotime Institute of Biotechnology). The concentration of the total protein extracted from each group was measured via the BCA method, according to the manufacturer's instructions. The proteins (15 µl/lane; containing 25–40 µg of protein) were then denatured and separated in a 10% sodium dodecyl sulfate-polyacrylamide gel and then transferred to a polyvinylidene fluoride membrane. The membrane was blocked with 5% skimmed milk for 2 h at room temperature and then incubated with rabbit anti-SENP1 (1:2,000), rabbit anti-HIF-1α (1:2,000), rabbit anti-GAPDH (1:3,000), rabbit anti-HO-1 (1:5,000), rabbit anti-Bax (1:1,000), rabbit anti-Bcl-2 (1:1,000), rabbit anti-β-actin (1:3,000) and rabbit anti-cleaved-caspase-3 (1:1,000) overnight at 4°C. After washing, a goat anti-rabbit HRP antibody (1:2,000) was incubated with the membrane for 1 h at 37°C. Finally, the protein bands were detected by the enhanced chemiluminescence method (Life-ilab), and the relative protein levels were determined by scanning densitometry analysis in Quantity One software (version 4.6; Bio-Rad Laboratories, Inc.) with GAPDH and β-actin as the loading controls.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
After the cells were treated as aforementioned, total RNA was extracted using TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. RT of ~2 µg of RNA into cDNA was performed using a Prime Script RT kit (TransGen Biotech Co., Ltd.) according to the manufacturer's protocol. qPCR analysis was then performed with a Light Cycler Real-Time PCR System (LightCycler 480; Roche Diagnostics; SYBR Green). The amplification procedure was as follows: 95°C for 30 sec, 40 cycles at 95°C for 5 sec, 60°C for 30 sec and 72°C for 60 sec. The results were determined using the 2−ΔΔCq method (21) and are expressed as the fold difference relative to the GAPDH level. The primers used are shown in Table I.
Enzyme-linked immunosorbent assays (ELISAs)
The supernatant of the cultured cells was collected, and the levels of VEGF (cat. no. MMV00), IL-6 (cat. no. M6000B) and TNF-α (cat. no. MTA00B) were determined with commercial ELISA kits (R&D Systems, Inc.). The sample was diluted five times, and all the experiments were conducted according to the manufacturer's instructions. Each experiment was repeated three times.
Statistical analysis
All data are presented as the average ± standard deviation. The P-values were determined by Ordinary one-way ANOVA (Tukey's multiple comparisons test), two-way ANOVA (Sidak's multiple comparisons test) and unpaired t-tests. All analyses were performed using Prism 8 (Dotmatics). P<0.05 was considered to indicate a statistically significant difference.
Results
Expression of SENP1 in LSECs is significantly upregulated following H-R
After the H-R LSEC model was established, it was verified via RT-qPCR that the level of SENP1 mRNA in these LSECs was significantly higher than that in the control (normoxic) cells (P<0.001; Fig. 1C). Similarly, it was demonstrated by WB that the protein expression level of SENP1 in the H-R LSEC model was significantly higher than that in the control (normoxic) cells (P<0.0001; Fig. 1A and B). From these results, it can be suggested that SENP1 may be involved in the regulation of H-R injury in LSECs.
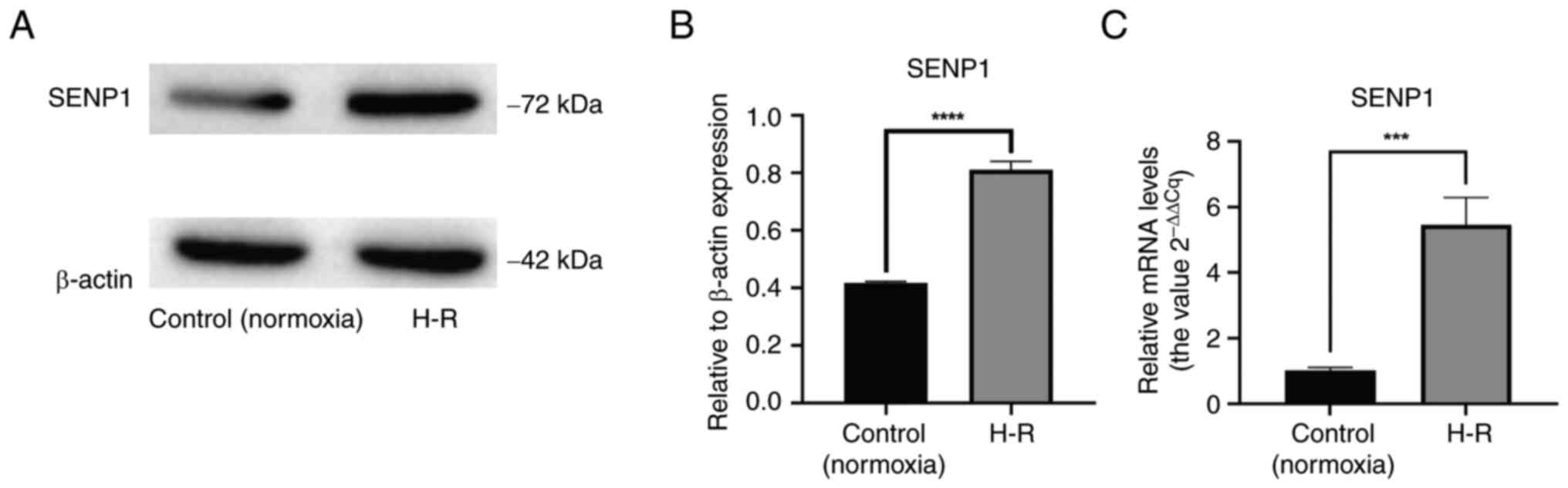
Viability of LSECs is impaired and fenestration damage is increased following H-R
To explore the phenotypic changes in primary mouse LSECs following H-R injury, a CCK-8 assay was conducted to measure cell viability. It was found that, compared with the control (normoxic) group, the viability of the H-R injured LSECs decreased significantly (P<0.0001; Fig. 2C).
To explore the effect of H-R on LSEC fenestration, scanning electron microscopy was performed to identify fenestrated cells. Compared with the control (normoxic) group, the fenestration in the H-R injured cells was significantly damaged, with a significant decrease in the number of fenestrated cells (P<0.05; Fig. 2A and B). These results suggested that the fenestration of LSECs decreased rapidly following H-R, which accelerated cell dedifferentiation.
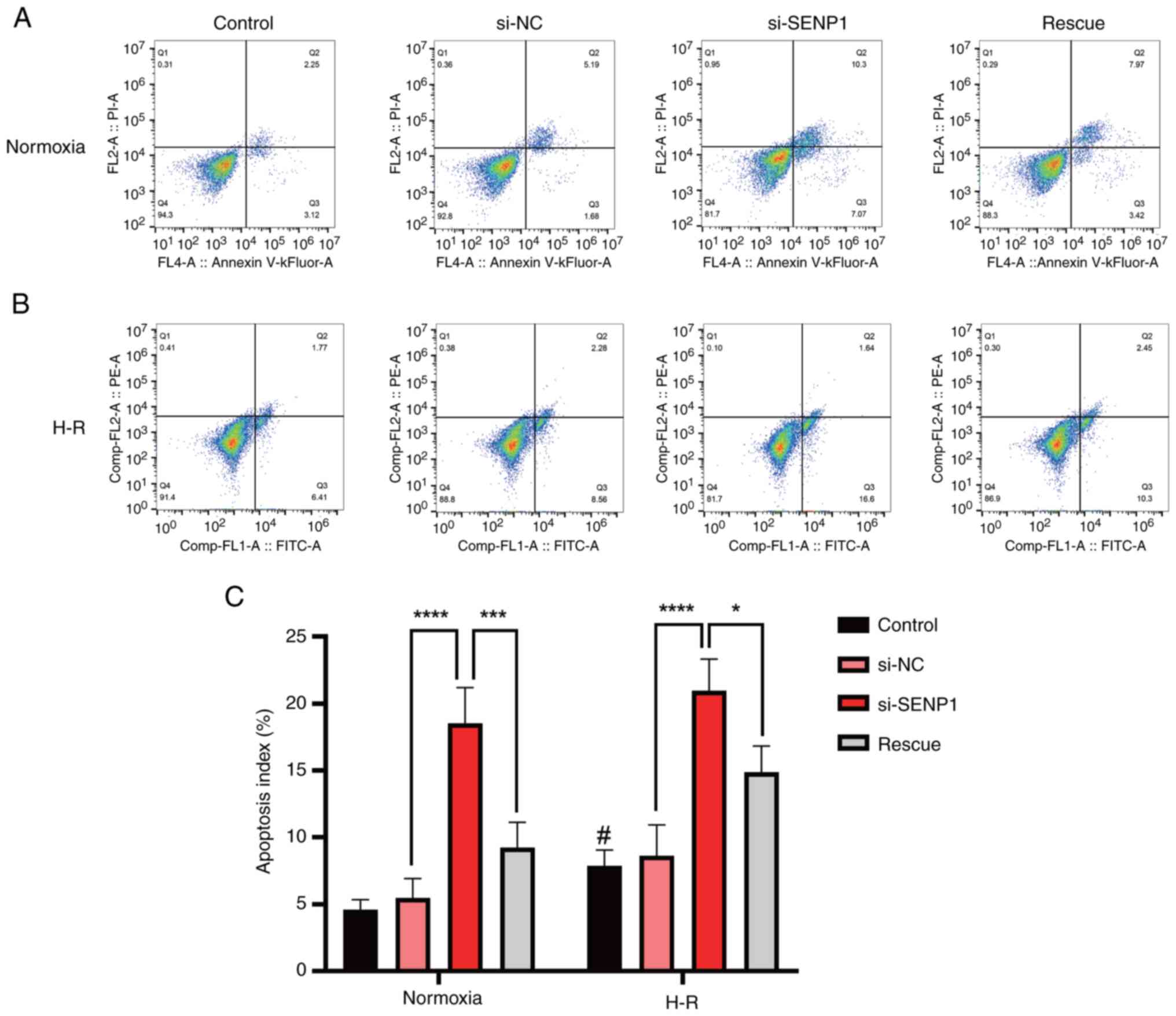
Apoptosis rate of LSECs is significantly increased following H-R
To explore the effect of H-R injury on primary mouse LSEC apoptosis, the number of apoptotic cells was measured by flow cytometry. It was found that the apoptosis rate of the H-R injured cells was significantly higher than that of the control (normoxic) group (Fig. 3A-C).
Silencing of SENP1 in H-R injured LSECs decreases the proliferation rate and increases the extent of fenestration damage and the apoptosis rate
To determine whether the phenotypic changes in LSECs following H-R injury, such as the decrease in proliferation rate and fenestration and the increase in apoptosis, were related to changes in SENP1 protein expression, the SENP1 expression in LSECs was silenced with siRNA. It was found that, compared with the control group, after silencing SENP1 expression, the decrease in the proliferation rate, the extent of diminished fenestration and the increase in the apoptosis rate were all exacerbated. This indicated that SENP1 may be involved in the regulation of LSEC proliferation, fenestration and apoptosis after H-R exposure (Figs. 2 and 3).
Following activation of the HIF-1α signaling pathway, the reduction in LSEC fenestration and proliferation rate is alleviated and the apoptosis rate is decreased
After activating the HIF-1α signaling pathway with the HIF-1α agonist, DMOG, the reduction in LSEC fenestration and proliferation rate was significantly alleviated and the apoptosis rate was significantly decreased (Figs. 2 and 3). These results suggested that SENP1 may regulate LSEC fenestration maintenance, proliferation and apoptosis after H-R exposure through the HIF-1α signaling pathway.
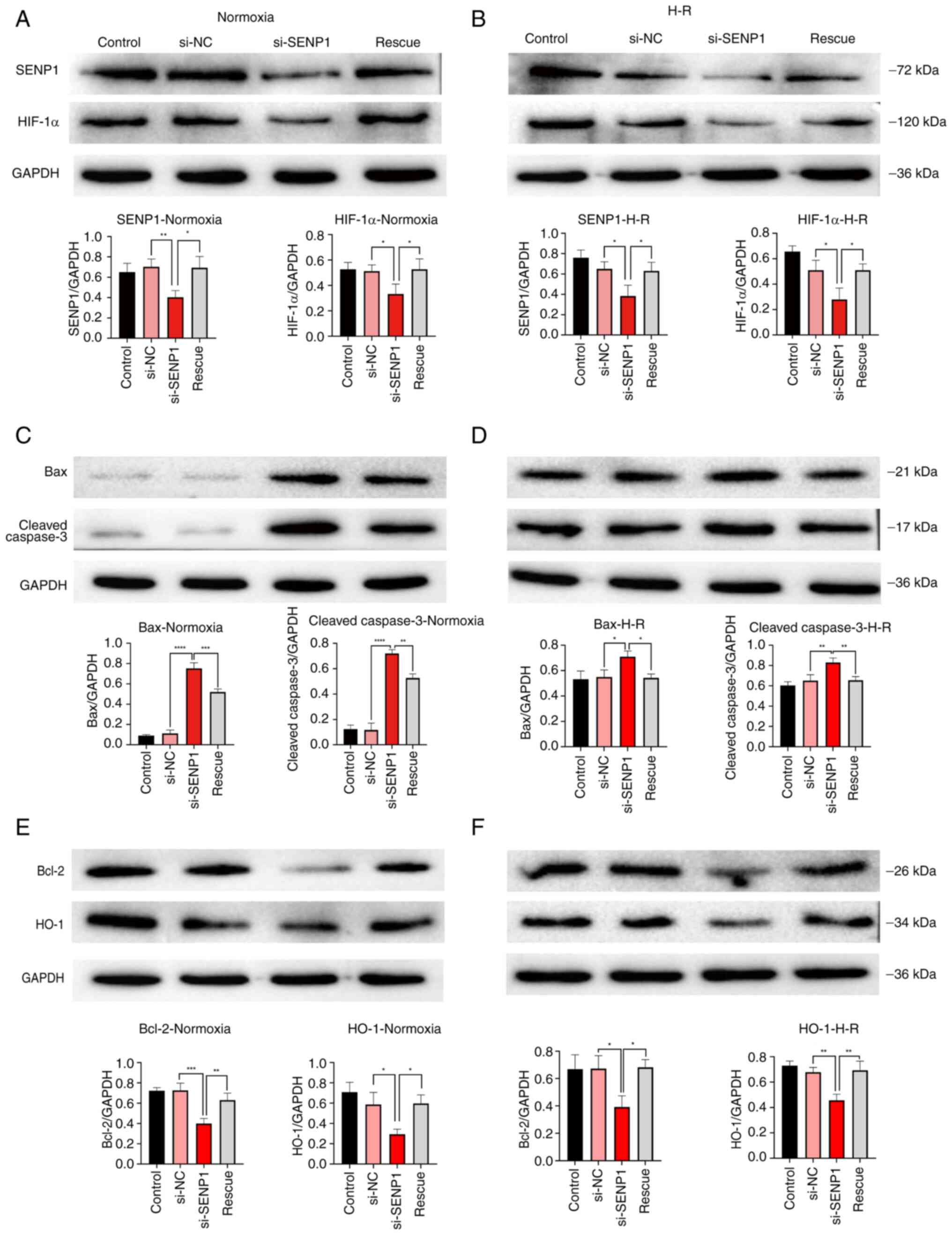
SENP1 attenuates the decrease in proliferation rate, increase in apoptosis rate and damage to fenestration in H-R injured LSECs through the HIF-1α signaling pathway
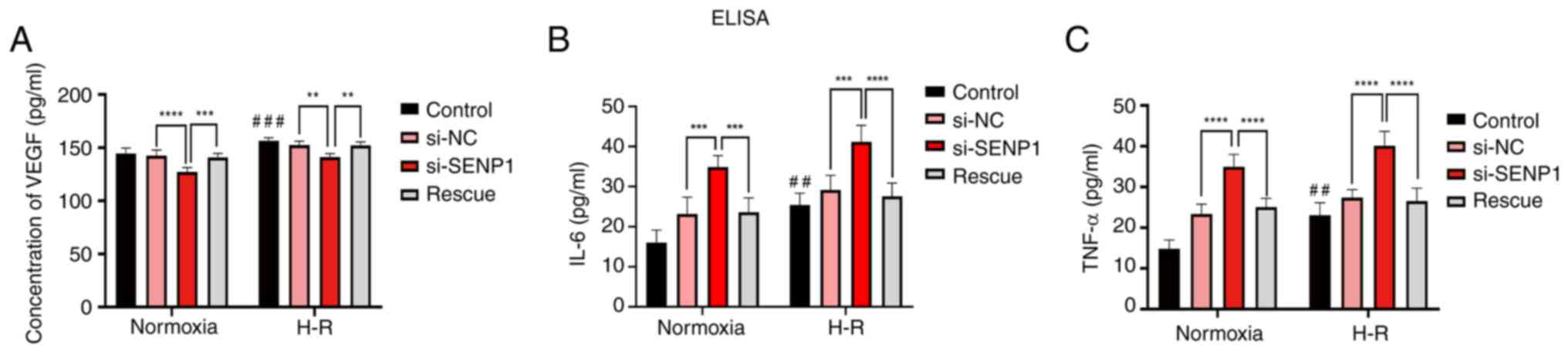
To further explore the specific mechanism by which SENP1 regulated the proliferation, apoptosis and fenestration of LSECs following H-R injury, SENP1 expression in LSECs was silenced with siRNA. It was found that si-SENP1 not only decreased the SENP1 protein level but also decreased the expression of HIF-1α and HO-1, increased the expression of apoptosis-related proteins, cleaved-Caspase-3 and Bax, and decreased the expression of Bcl-2 (Fig. 4A-F). In addition, the levels of VEGF, IL-6 and TNF-α in the cell culture medium were determined by ELISA (Fig. 5). The levels of IL-6 and TNF-α in the H-R group were significantly higher than those in the control (normoxic) group. The concentration of VEGF was also increased (Fig. 5). However, compared with the si-NC group, the VEGF protein expression level in the si-SENP1 group decreased significantly, while the levels of IL-6 and TNF-α increased significantly (Fig. 5). However, after the HIF-1α signaling pathway agonist, DMOG, was added to cells (the rescue group), the results were the opposite of that following SENP1 silencing. In addition, the HIF-1α signaling pathway was reactivated, attenuating the damage caused by H-R. This indicated that silencing SENP1 may inhibit the effect of the HIF-1α signaling pathway and subsequently aggravate the damage caused by H-R. Specifically, these results suggested that SENP1 attenuated the increase in apoptosis and fenestration impairments in LSECs following H-R through the HIF-1α signaling pathway.
Discussion
Hepatic I/R injury is a common pathophysiological outcome of liver transplantation and hepatectomy (1,2). LSECs experience H-R injury, which proceeds through two different stages (1,2). During the ischemic period, blood flow to the liver is interrupted, resulting in tissue hypoxia, which damages the normal function of mitochondria (1,2,22). Specifically, the lack of oxygen delivered via electron carriers to the end of the mitochondrial respiratory chain immediately interrupts electron flow, resulting in reduced respiratory chain output (22). The subsequent interruption to oxidative phosphorylation leads to the rapid depletion of ATP, the acceleration of glycolysis, an increase in lactic acid production and a change in Ca2+ homeostasis, which all exert damaging effects on LSECs, hepatocytes and other liver cells (23). Although blood flow to the liver is restored through reperfusion, the number of inflammatory cells, pro-inflammatory mediators and reactive oxygen species increases, exacerbating liver injury (24). Furthermore, damage to LSECs leads to a lack of neovascularization in hyperplastic liver tissue and the disruption of microcirculation in hepatic sinusoids, exacerbating ischemic and hypoxic damage to hepatocytes and other non-parenchymal cells (6). These outcomes increase the hepatocyte apoptosis and necrosis rates, eventually aggravating hepatic I/R injury and potentially inducing liver failure (6). Therefore, reducing H-R injury to LSECs would increase the stability of microcirculation in the hepatic sinusoid environment and alleviate I/R injury.
Previous studies have shown that LSECs not only coordinate the response of the liver to injury but also mediate the recovery of liver injury (2,12,13). LSECs reduce the effect of I/R injury through autophagy, thus ensuring normal cell function (25). Furthermore, LSECs with normal function ensure liver regeneration and liver function recovery following hepatectomy (12,13). A number of mechanisms may regulate the activation of autophagy in endothelial cells. A decrease in ATP or in the availability of growth factors leads to the activation of AMP-activated protein kinase (AMPK). Once activated, AMPK inhibits mTOR action, resulting in the activation of unc-51 like autophagy activating kinase (ULK1), which triggers autophagy. In endothelial cells, calmodulin-dependent protein kinase-β-mediated AMPK activation inhibits mTOR activity, resulting in the upregulation of ULK1 and autophagy activation. Therefore, in endothelial cells, the activation of autophagy involves the dynamic interaction between AMPK, intracellular calcium, mTOR and ULK-1 (26,27). However, when H-R injury is severe, LSECs show dysfunction, the hepatic sinusoid microenvironment is subjected to ischemic and hypoxic conditions, the number of inflammatory mediators increases and reactive oxygen species accumulate (1,6,22). The LSECs and hepatocytes then undergo necrosis or apoptosis and liver regeneration is attenuated, making liver failure a likely outcome (1,22). Therefore, reducing the damage to LSECs caused by I/R injury promotes liver regeneration and liver function recovery following hepatectomy.
Our previous study revealed that, under hypoxic culture conditions, LSECs maintained fenestration and proliferated via regulation of the SENP1/HIF-1α/VEGF signaling pathway (17,28). Thus, SENP1 has been suggested to be involved in regulating the maintenance of a healthy LSEC phenotype under hypoxic conditions, assisting LSEC function in promoting liver regeneration. However, during and after hepatectomy, the pathophysiological process of I/R in hepatic sinusoids due to the disappearance of a hepatic artery buffering effect and unbalanced proliferation of cells in the liver during liver regeneration, leads to H-R injury to LSECs (29). The expression of SENP1 has been reported to be upregulated in cardiomyocytes during H-R injury, reducing H-R injury (30). Our previous study confirmed that SENP1 expression was significantly upregulated in hypoxia-treated LSECs and participated in the regulation of LSEC proliferation and fenestration maintenance. Therefore, it was hypothesized that SENP1 may be involved in regulating the pathological process underlying H-R injury in LSECs.
In the present study, using a mouse primary LSEC H-R model, it was found that SENP1 expression in LSECs was significantly upregulated at both the mRNA and protein levels compared with the control group. These findings therefore indicated that SENP1 expression in LSECs is affected by H-R. In addition, it was found that the proliferation rate was reduced, fenestration damage was increased and the apoptosis rate was increased in LSECs cultured in H-R conditions. To explore whether these phenotypic changes were related to changes in SENP1 expression, siRNA was used to silence SENP1 expression in LSECs. It was demonstrated that, compared with the control group, the reduction in viability was more profound, fenestration damage was increased and the apoptosis rate was increased in cells transfected with si-SENP1. This indicated that SENP1 may be involved in the regulation of viability, fenestration and apoptosis in LSECs exposed to H-R.
To further explore the specific mechanisms by which SENP1 regulates the viability, apoptosis and fenestration of LSECs following H-R, the expression levels of associated factors were assessed. It was found that si-SENP1 transfection not only decreased the protein level of SENP1 but also decreased the expression of HIF-1α and HO-1, increased the expression of the apoptosis-related proteins, caspase-3 and Bax, and decreased the expression of Bcl-2. These results therefore revealed that silencing SENP1 expression suppressed the HIF-1α signaling pathway, increasing the activity of the intrinsic apoptosis pathway, and thus increasing LSEC apoptosis. In addition, ELISAs were performed to measure the levels of VEGF, IL-6 and TNF-α in the cell culture medium. It was found that, compared with the control group, the levels of VEGF, IL-6 and TNF-α were significantly increased in the H-R group. However, compared with the H-R group, the protein expression of VEGF was decreased, while the levels of IL-6 and TNF-α were significantly increased in the group transfected with si-SENP1. Studies (12–15) have demonstrated that VEGF participated in maintaining LSEC differentiation and fenestration. However, when LSECs were damaged by H-R, the expression of VEGF increased, delaying the loss of fenestration. Moreover, after SENP1 silencing, the expression of VEGF decreased significantly, and the loss of fenestration was more notable, indicating that SENP1 regulated the expression of VEGF in LSECs, which was similar to the findings of our previous study (17). In the present study, IL-6, TNF-α and other inflammatory mediator levels were found to be increased in LSECs following H-R exposure, which was similar to previous findings reported in the literature (20,31). These findings suggested that the number of LSECs may increase due to the increase in inflammatory factors that trigger the intrinsic apoptosis pathway, thus explaining the increase in the LSEC apoptosis rate following H-R injury.
However, the limitations of the present study should be acknowledged. The experiments were conducted in vitro and cannot truly reflect the complex internal environment in vivo. Studies have shown that in vivo, VEGF is secreted mainly by hepatocytes and hepatic stellate cells, which affect LSECs through paracrine signaling (13,15). The results of the present study demonstrated that LSECs self-regulate fenestration via autocrine VEGF.
In addition, through the use of an HIF-1α signaling pathway agonist, the outcomes of the aforementioned experiments were reversed. Specifically, the inhibitory effect of si-SENP1 was counteracted, indicating that silencing SENP1 expression may inhibit the effect of the HIF-1α signaling pathway, aggravating the damage caused by H-R. However, when a HIF-1α signaling pathway agonist was used, the damage caused by H-R was alleviated. Therefore, SENP1 may reduce the apoptosis rate and fenestration damage in LSECs following H-R through the HIF-1α signaling pathway.
In summary, after H-R treatment, the proliferation rate was decreased, fenestration was inhibited, the apoptosis rate increased and SENP1 expression was increased in LESCs. Moreover, after SENP1 silencing, the expression of HIF-1α decreased and the damage to LSECs was aggravated. However, the H-R injury of LSECs was reversed when an HIF-1α signaling pathway agonist was used to treat the cells. These findings indicated that SENP1 may alleviate H-R injury to LSECs by mediating the effects of HIF-1α signaling.
Acknowledgements
Not applicable.
Funding
This research was supported by the Scientific Research Foundation Project of the Department of Science and Technology of Yunnan Province (grant no. 202101AY070001-128) and the National Natural Science Foundation of China (grant no. 81960124).
Availability of data and materials
The data generated in the present study may be requested from the corresponding author.
Authors' contributions
ZQ, QL and ZZ designed the study. ZQ and QL performed most of the data analysis, imaging and cell experiments. QL and JD performed all scanning electron microscopy experiments. QL performed the cell experiments. JD, JL, SY and HH collected and analyzed the data. JL, SY and HH performed literature review. ZQ and QL wrote the manuscript. ZZ and HH revised the manuscript. All authors read and approved the final version of the manuscript. ZQ, QL and ZZ confirm the authenticity of all the raw data.
Ethics approval and consent to participate
All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Ethics Committee of the First Affiliated Hospital of Kunming Medical University (Kunming, China; approval no. KMMU2020202).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Glossary
Abbreviations
Abbreviations:
|
LSECs |
liver sinusoidal endothelial cells |
|
SENP1 |
Sentrin/SUMO-specific protease 1 |
|
HIF-1α |
hypoxia-inducible transcription factor-1α |
|
VEGF |
vascular endothelial growth factor |
|
I/R |
ischemia-reperfusion |
|
H-R |
hypoxia-reoxygenation |
|
RT-qPCR |
reverse transcription-quantitative polymerase chain reaction |
|
WB |
western blotting |
|
ELISA |
enzyme-linked immunosorbent assay |
References
|
Yang W, Chen J, Meng Y, Chen Z and Yang J: Novel targets for treating ischemia-reperfusion injury in the liver. Int J Mol Sci. 19:13022018. View Article : Google Scholar : PubMed/NCBI | |
|
Selzner N, Rudiger H, Graf R and Clavien PA: Protective strategies against ischemic injury of the liver. Gastroenterology. 125:917–936. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Dar WA, Sullivan E, Bynon JS, Eltzschig H and Ju C: Ischaemia reperfusion injury in liver transplantation: Cellular and molecular mechanisms. Liver Int. 39:788–801. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Russo L, Gracia-Sancho J, García-Calderó H, Marrone G, García-Pagán JC, García-Cardeña G and Bosch J: Addition of simvastatin to cold storage solution prevents endothelial dysfunction in explanted rat livers. Hepatology. 55:921–930. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Peralta C, Jiménez-Castro MB and Gracia-Sancho J: Hepatic ischemia and reperfusion injury: Effects on the liver sinusoidal milieu. J Hepatol. 59:1094–1106. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Hide D, Warren A, Fernandez-Iglesias A, Maeso-Diaz R, Peralta C, Le Couteur DG, Bosch J, Cogger VC and Gracia-Sancho J: Ischemia/reperfusion injury in the aged liver: The importance of the sinusoidal endothelium in developing therapeutic strategies for the elderly. J Gerontol A Biol Sci Med Sci. 75:268–277. 2020.PubMed/NCBI | |
|
Wang X, Walkey CJ, Maretti-Mira AC, Wang L, Johnson DL and DeLeve LD: Susceptibility of Rat steatotic liver to ischemia-reperfusion is treatable with liver-selective matrix metalloproteinase inhibition. Hepatology. 72:1771–1785. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Stolz DB, Ross MA, Ikeda A, Tomiyama K, Kaizu T, Geller DA and Murase N: Sinusoidal endothelial cell repopulation following ischemia/reperfusion injury in rat liver transplantation. Hepatology. 46:1464–1475. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Caldwell-Kenkel JC, Currin RT, Tanaka Y, Thurman RG and Lemasters JJ: Reperfusion injury to endothelial cells following cold ischemic storage of rat livers. Hepatology. 10:292–299. 1989. View Article : Google Scholar : PubMed/NCBI | |
|
Hui AM, Kawasaki S, Makuuchi M, Nakayama J, Ikegami T and Miyagawa S: Liver injury following normothermic ischemia in steatotic rat liver. Hepatology. 20:1287–1293. 1994. View Article : Google Scholar : PubMed/NCBI | |
|
Jawad R, D'Souza M, Selenius LA, Lundgren MW, Danielsson O, Nowak G, Björnstedt M and Isaksson B: Morphological alterations and redox changes associated with hepatic warm ischemia-reperfusion injury. World J Hepatol. 9:1261–1269. 2007. View Article : Google Scholar | |
|
DeLeve LD: Liver sinusoidal endothelial cells and liver regeneration. J Clin Invest. 123:1861–1866. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
DeLeve LD, Wang X, Hu L, McCuskey MK and McCuskey RS: Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol Gastrointest Liver Physiol. 287:G757–G763. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
DeLeve LD and Maretti-Mira AC: Liver sinusoidal endothelial cell: An update. Semin Liver Dis. 37:377–387. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Funyu J, Mochida S, Inao M, Matsui A and Fujiwara K: VEGF can act as vascular permeability factor in the hepatic sinusoids through upregulation of porosity of endothelial cells. Biochem Biophys Res Commun. 280:481–485. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, Gaarde WA and Deleve LD: Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology. 142:918–927.e6. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Qing Z, Huang H, Yang S, Lin J, Zeng Z, Duan J, Yuan B and Ming T: Hypoxia maintains the fenestration of liver sinusoidal endothelial cells and promotes their proliferation through the SENP1/HIF-1α/VEGF signaling axis. Biochem Biophys Res Commun. 540:42–50. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Cheng J, Kang X, Zhang S and Yeh ET: SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 131:584–695. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Cui CP, Wong CCL, Kai AKL, Ho DWH, Lau EYT, Tsui YM, Chan LK, Cheung TT, Chok KSH, Chan ACY, et al: SENP1 promotes hypoxia-induced cancer stemness by HIF-1α deSUMOylation and SENP1/HIF-1α positive feedback loop. Gut. 66:2149–2159. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Qu S, Yuan B, Zhang H, Huang H, Zeng Z, Yang S, Ling J, Jin L and Wu P: Heme oxygenase 1 attenuates hypoxia-reoxygenation injury in mice liver sinusoidal endothelial cells. Transplantation. 102:426–432. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Saidi RF and Kenari SKH: Liver ischemia/reperfusion injury: An overview. J Invest Surg. 27:366–379. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Gasbarrini A, Borle AB, Farghali H, Bender C, Francavilla A and Van Thiel D: Effect of anoxia on intracellular ATP, Na+i, Ca2+i, Mg2+i, and cytotoxicity in rat hepatocytes. J Biol Chem. 267:6654–6663. 1992. View Article : Google Scholar : PubMed/NCBI | |
|
Cannistrà M, Ruggiero M, Zullo A, Gallelli G, Serafini S, Maria M, Naso A, Grande R, Serra R and Nardo B: Hepatic ischemia reperfusion injury: A systematic review of literature and the role of current drugs and biomarkers. Int J Surg. 33 (Suppl 1):S57–S70. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Høyer-Hansen M and Jäättelä M: AMP-activated protein kinase: A universal regulator of autophagy? Autophagy. 3:381–383. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Kim HS, Montana V, Jang HJ, Parpura V and Kim JA: Epigallocatechin gallate (EGCG) stimulates autophagy in vascular endothelial cells: A potential role for reducing lipid accumulation. J Biol Chem. 288:22693–22705. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Ghislat G, Patron M, Rizzuto R and Knecht E: Withdrawal of essential amino acids increases autophagy by a pathway involving Ca2+/calmodulin-dependent kinase kinase-β (CaMKK-β). J Biol Chem. 287:38625–38636. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Qing Z, Huang H, Luo Q, Lin J, Yang S, Liu T, Zeng Z and Ming T: Hypoxia promotes the proliferation of mouse liver sinusoidal endothelial cells: miRNA-mRNA expression analysis. Bioengineered. 12:8666–8678. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D and Rautou PE: Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol. 66:212–227. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Gu J, Fan Y, Liu X, Zhou L, Cheng J, Cai R and Xue S: SENP1 protects against myocardial ischaemia/reperfusion injury via a HIF1α-dependent pathway. Cardiovasc Res. 104:83–92. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Pibiri M, Leoni VP and Atzori L: Heme oxygenase-1 inhibitor tin-protoporphyrin improves liver regeneration after partial hepatectomy. Life Sci. 204:9–14. 2018. View Article : Google Scholar : PubMed/NCBI |