Bif‑1 inhibits activation of inflammasome through autophagy regulatory mechanism
- Authors:
- Published online on: March 1, 2024 https://doi.org/10.3892/mmr.2024.13191
- Article Number: 67
-
Copyright: © Zhang et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Inflammatory reactions are the key to the occurrence and treatment of various diseases, such as sepsis and cardiovascular disorders (1,2). Among the complex mechanisms of the inflammation process, there is an important mechanism related to the nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing protein 3 (NLRP3) inflammasome, the activation of which often acts as the key mechanism in the inflammatory response (3). Signal 1 is triggered by the activation of pathogen-associated molecular patterns (PAMPs). This activation leads to the upregulation of NLRP3 inflammasome transcription, which is dependent on the nuclear factor-κB (NF-κB) pathway (4). NF-κB is a family of transcription factors involved in regulating various biological processes, including inflammasome activation and cell survival (5). When inflammatory signals are activated, the phosphorylated form of p65 is released and enters the nucleus, binds to target genes and regulates their transcriptional activities, thus regulating the extent and duration of the inflammatory response (6). Signal 1 and 2 can be triggered by PAMPs; signal 2 is also regulated by other damage-associated molecular patterns (DAMPs) such as adenosine triphosphate (ATP), a classical DAMP, typically leading to the initiation of various upstream signaling events (7). During the inflammation process, the activated NLRP3 inflammasome recruits the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) to facilitate the nucleation of ASC filaments, which serves as a platform for the recruitment and activation of caspase-1 (8). This activation of caspase-1 leads to the processing and release of proinflammatory cytokines and can trigger pyroptotic cell death (9). Upon activation, caspase-1 cleaves pro-interleukin (IL)-1β and pro-IL-18, leading to the generation of their active forms (10). The active form of IL-1β plays a critical role in promoting inflammation and regulating cellular immune responses by recruiting and activating immune cells (11).
Autophagy maintains the homeostasis of the intracellular environment by removing damaged organelles and metabolites, thereby inhibiting the onset and progression of inflammation (12,13). Autophagy can inhibit inflammasome formation and IL-1β production by degrading components of the NLRP3 inflammasome, such as NLRP3, ASC and pro-IL-1β (14,15). Lipopolysaccharide (LPS) treatment stimulates toll-like receptor 4 on the surface of macrophages, which in turn activates autophagy-related signaling pathways (16). p62, a classical autophagy receptor, is involved in autophagosome formation (17). In addition, the conversion of microtubule-associated protein 1 light chain 3 (LC3) to the LC3-II form has been widely used to assess autophagic activity (18).
Bax-interacting factor 1 (Bif-1), also known as SH3GLB1 or lipophilic enzyme B1, is present in the cytoplasm, and is a key protein in the process of autophagy and autophagosome formation (19–21). The loss of Bif-1 may impede the endogenous apoptotic pathway while facilitating tumor development (22,23). Bif-1 exerts inhibitory effects on the production of mitochondrial and glycolytic ATP, and its downregulation contributes to the proliferation of melanoma cells (24). However, there is also contradictory evidence that Bif-1 is involved in tumor formation to a certain extent (25). In addition, studies have shown that Bif-1 is crucial for maintaining the morphology and function of mitochondria (26,27). However, the specific mechanism and the role of Bif-1 in the development of inflammation is still unclear.
In the present study, the critical role of Bif-1-mediated regulation of autophagy in inflammasome activation and inflammatory factor release was revealed. These findings provide new insights in the understanding of the regulatory mechanisms of the inflammatory response.
Materials and methods
Cell culture
J77A4A.1 (cat. no. CL-0370) were provided by Procell Life Science & Technology Co., Ltd. The cells were cultured in RPMI 1640 medium (Gibco; Thermo Fisher Scientific, Inc.) with 10% fetal bovine serum (Premium; PAN-Biotech GmbH) and 1% penicillin-streptomycin antibiotic (Gibco; Thermo Fisher Scientific, Inc.) at 37°C. To induce inflammation, cells were first primed with LPS (500 ng/ml; Sigma-Aldrich; Merck KGaA) for 6 h at 37°C. All inhibitors used in the present study were added at the same time as LPS. Specifically, the following inhibitors were employed: MLN120B (1 µM; Sigma-Aldrich; Merck KGaA) and MRT68921 (0.25 µM; MedChemExpress). Subsequently, cells were stimulated with ATP (5 mM; Sigma-Aldrich; Merck KGaA) for 30 min at 37°C.
After concentration in ultrafiltration centrifuge tubes (3 kDa; Merck KGaA), the supernatant was collected. Cells were washed twice with ice-cold phosphate-buffered saline (PBS; Sangon Biotech Co., Ltd.). For RNA extraction, 1 ml RNA isolation reagent (TRIzol®; Invitrogen; Thermo Fisher Scientific, Inc.) was added to the cells. For protein extraction, 70 µl lysis buffer [50 mM Tris (Sangon Biotech Co., Ltd.), 150 mM NaCl (Sangon Biotech Co., Ltd.), 0.1% (w/v) sodium dodecyl sulfate (Sangon Biotech Co., Ltd.), 1% Triton X-100 (Sangon Biotech Co., Ltd.) and one tablet/50 ml Protease Inhibitor Cocktail (Roche Diagnostics); pH 8.0] was added.
Animal experiments
Male NIH Swiss mice, aged 4 weeks, with an average weight of ~18 g, were obtained from Guangdong Medical Laboratory Animal Center (derived from National Institutes of Health). They were housed in controlled conditions with a constant temperature of 22±2°C, humidity of 60±5% and a 12 h dark/light cycle. The study strictly adhered to the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. The protocol was approved by The Bioethics Committee of the Shenzhen International Graduate School, Tsinghua University [Shenzhen, China; Ethical issue (2020) No. 9]. The mice were divided into either the control group or the LPS (10 mg/kg i.p.; Sigma-Aldrich; Merck KGaA) group, with each group consisting of three mice. The mice had ad libitum access to food and water throughout the experiment.
After a 4 h period of LPS induction, the mice were anesthetized with an intraperitoneal injection of a 10% (w/v) urethane solution (10 ml/kg; dissolved in normal saline; Sangon Biotech Co., Ltd.), equivalent to a dose of 1,000 mg/kg. Blood samples were collected and stored at −80°C for further research. Finally, the mice were euthanized by cervical dislocation.
siRNA transfection
siRNA transfection was performed as described in our previous study, and successful knockdown was confirmed by western blotting analysis (28). J774A.1 cells were cultured in 6-well plates until they reached 50% confluence. For every well, a transfection mixture was prepared, consisting of 250 µl transfection reagent [5 µl Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.) and 245 µl Opti-MEM (Gibco; Thermo Fisher Scientific, Inc.)], and 250 µl siRNA solution (100 nM; si-Sh3glb1; siB119191002-1-5; Guangzhou RiboBio Co., Ltd.; diluted with Opti-MEM). The siRNA sequence targeting Bif-1 was as follows: 5′-GGGCAAGGTGCCAATTACCTACTTA-3′. For the siRNA negative control, the sequence used was: 5′-CAGUACUUUUGUGUAGUACAA-3′ (100 nM; Guangzhou RiboBio Co., Ltd.; diluted with Opti-MEM). These mixed solutions were incubated for 20 min at room temperature. Next, cells were washed twice with PBS, and 1.5 ml Opti-MEM along with the mixed solution were added to every well. A total of 24 h after transfection, cells were primed with LPS (500 ng/ml) for 6 h, followed by stimulation with 5 mM ATP for 30 min. The incubations were performed at a constant temperature of 37°C to maintain physiological conditions.
Western blotting
The Bradford assay was performed using the Bradford protein concentration determination kit (cat. no. P0006; Beyotime Institute of Biotechnology) according to the manufacturer's instructions. For each lane, an equal mass of protein (25 µg) was loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels (12.5%; Epizyme Biomedical Technology Co., Ltd). The protein samples were subsequently transferred to nitrocellulose membranes (BioTrace, Ltd.). Membranes were blocked using a solution of 5% (w/v) non-fat milk (Anchor Ltd.) dissolved in tris-buffered saline with 0.1% Tween 20 (TBST) for 2 h at room temperature. After blocking, the membranes were incubated with primary antibodies overnight at 4°C. The primary antibodies were dissolved in 1.5% bovine serum albumin (BioFroxx; neoFroxx GmbH). Following three washes with TBST, the membranes were incubated with the corresponding secondary antibodies for 1 h at room temperature and then washed again with TBST. To visualize the protein bands, an enhanced chemiluminescence solution (Thermo Fisher Scientific, Inc.) was used. Using ImageJ (version 1.45; National Institutes of Health), the relative grey density values of the protein bands were quantified and subsequently normalized to the density of β-actin. The specific primary antibodies used in the present study included anti-β-actin (1:50,000; cat. no. A1978; Sigma-Aldrich; Merck KGaA), anti-Bif-1 (1:1,000; cat. no. 4467S; Cell Signaling Technology, Inc.), anti-NF-κB p65 (1:2,000; cat. no. 6956S; Cell Signaling Technology, Inc.), anti-phospho-NF-κB p65 (1:2,000; cat. no. 3033S; Cell Signaling Technology, Inc.), anti-NLRP3 (1:1,000; cat. no. 15101S; Cell Signaling Technology, Inc.), anti-IL-1β (1:1,000; cat. no. 12242S; Cell Signaling Technology, Inc.), anti-p62 (1:2,000; cat. no. 88588S; Cell Signaling Technology, Inc.) and anti-LC3B (1:2,000; cat. no. A19665; ABclonal Biotech Co., Ltd.). The secondary antibodies used included goat polyclonal antibody to rabbit IgG H&L HRP (1:5,000; cat. no. 7074; Cell Signaling Technology, Inc.) or goat polyclonal antibody to mouse IgG H&L HRP (1:5,000; cat. no. 32230; Thermo Fisher Scientific, Inc.).
ELISA
The supernatants from cell culture were collected and preserved at a temperature of −80°C. Subsequently, they were analyzed for IL-1β (cat. no. KET7005; Abbkine Scientific Co., Ltd.) levels following the manufacturer's instructions.
Reverse transcription-quantitative PCR (RT-qPCR)
RNAiso Plus (Takara Bio USA, Inc.) was used to extract total RNA from J774A.1 cells, following the manufacturer's instructions. The concentration of the extracted RNA was determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.). Subsequently, cDNA was synthesized using Evo M-MLV RT Premix (cat. no. AG11706; Accurate Biology). The kit included the reverse transcriptase enzyme, buffer, dNTPs, and primers necessary for cDNA synthesis. The reverse transcription protocol followed the manufacturer's instructions for the Evo M-MLV RT Premix kit (Hunan Accurate Bio-Medical Co., Ltd.), including temperature specifications. cDNA was quantitatively analyzed using the SYBR Green Premix Pro Taq HS qPCR Kit (Hunan Accurate Bio-Medical Co., Ltd.). Using the 2−∆∆Cq method, the relative gene expression was calculated with β-actin as the reference gene (29). The parameters required for denaturation, annealing and extension were as follows: 95°C for 30 sec, 40 cycles at 95°C for 5 sec and 60°C for 30 sec. The sequences of the primers used were: 5′-GCAACTGTTCCTGAACTCAACT-3′ and 5′-ATCTTTTGGGGTCCGTCAACT-3′ for IL-1β; 5′-ATTACCCGCCCGAGAAAGG-3′ and 5′-TCGCAGCAAAGATCCACACAG-3′ for Nlrp3; and 5′-GGCTGTATTCCCCTCCATCG-3′ and 5′-CCAGTTGGTAACAATGCCATGT-3′ for β-actin.
Immunofluorescence and confocal assay
The immunofluorescence assay in J774A.1 cells was performed following a previously described protocol (30). First, circular transparent glass slides (diameter, 10 mm) were placed at the bottom of a 6-well plate. A total of 2.5×105 cells/well (2 ml cell culture medium) were added to the surface of glass slides (cat. no. 12-545-83; Fisherbrand™ microscope cover glass; Thermo Fisher Scientific, Inc.) in a 6-well plate. The cells were cultured in fresh medium for 12 h and then stimulated with the corresponding drugs.
The immobilized cells on the slides were washed with PBS and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. The cells were then washed with PBS three times and incubated with 0.1% triton in PBS for 15 min. After three washes with PBS, cells were blocked with 3% bovine serum albumin (cat. no. A8010; Beijing Solarbio Science & Technology Co., Ltd.) in PBS for 1 h at room temperature. Next, cells were incubated with anti-LC3B (cat. no. A19665; ABclonal Biotech Co., Ltd.; 1:200) in PBS for 1 h at 37°C and washed with PBS three times.
The cells were incubated with goat anti-rabbit IgG H&L (1:1,000; cat. no. ab150077; Alexa Fluor® 488; Abcam) in PBS for 1 h at 37°C. The samples were sealed with Antifade mounting medium with DAPI (cat. no. P0131; Beyotime Institute of Biotechnology). The fluorescence signal was acquired using Nikon Confocal A1R system (Nikon Corporation).
Statistical analysis
The data are presented as mean ± SD of three independent experiments. Statistical comparisons between two groups were performed using one-way ANOVA followed by Tukey's post-hoc test. P<0.05 was considered to indicate a statistically significant difference. All statistical analyses and calculations were performed using GraphPad Prism (version 9.5; Dotmatics).
Results
Inflammasome is activated by the NF-κB pathway in LPS/ATP-induced J774A.1 cells
Previous studies have shown that LPS/ATP treatment can induce an inflammatory response (31,32). The inflammatory response is activated when the NLRP3 inflammasome senses signals from pathogenic microorganisms, cellular stress and injury. The transcription of inflammation-related genes and cellular immune responses is regulated by the NF-κB signaling pathway (33).
First, using the LPS/ATP-treated J774A.1 cell model, the levels of NLRP3 and IL-1β proteins were measured in cell lysates (Fig. 1A). The levels of NLRP3 and IL-1β proteins measured using western blotting were significantly increased in LPS/ATP-induced J774A.1 cells. Also, the level of IL-1β in the cell medium of LPS/ATP-induced J774A.1 cells was significantly increased as indicated by the results generated using ELISA (Fig. 1D), suggesting that LPS/ATP treatment can contribute to the activation of the NLRP3 inflammasome and lead to the release of IL-1β. This result is consistent with previous studies and supports the mechanism by which LPS/ATP treatment triggers an inflammatory response (34,35).
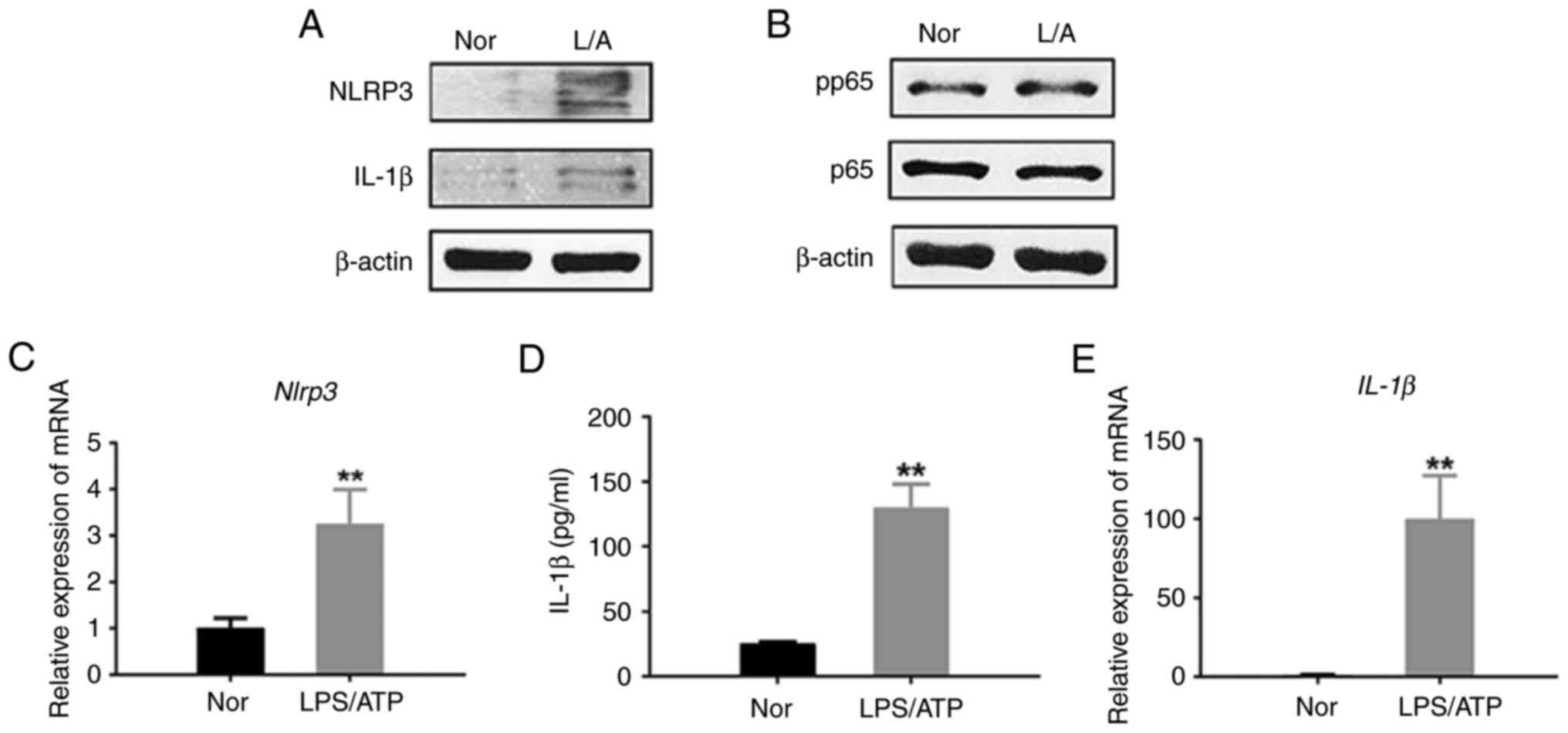
Secondly, there was a significant increase in the phosphorylation level of p65 in J774A.1 cells (Fig. 1B). Phosphorylated p65 is one of the hallmarks of NF-κB signaling pathway activation. Its activation significantly increased the mRNA levels of NLRP3 and IL-1β in J774A.1 cells as indicated by qPCR (Fig. 1C-E). Therefore, the results of the present study suggested that LPS/ATP treatment triggered the activation of the NF-κB signaling pathway, which may lead to increased transcription of inflammation-related genes.
si-Bif-1 significantly upregulates the IL-1β level in supernatants and lysates
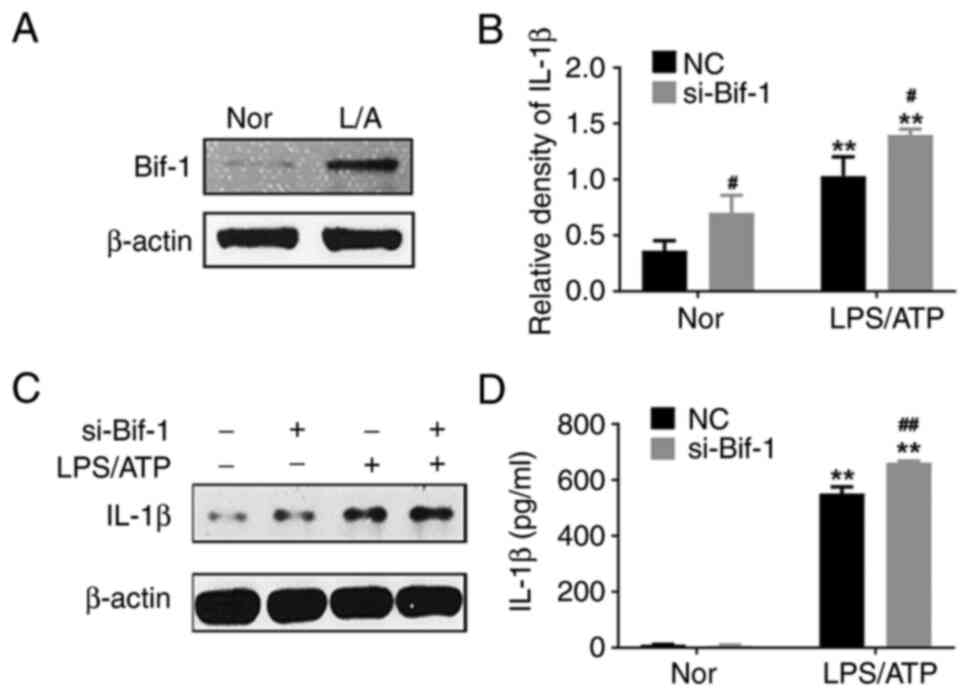
Bif-1 is a key factor in regulating autophagy. It was shown that Bif-1 was significantly increased in LPS/ATP-induced J774A.1 cells (Fig. 2A). The NIH mice were treated with LPS, as described in Fig. S1, and an increase in Bif-1 levels in peripheral blood mononuclear cells was observed. However, using si-Bif-1 in LPS/ATP-induced J774A.1 cells significantly increased IL-1β levels in supernatants and whole-cell lysates (Fig. 2B-D). It seemed that Bif-1 exerted an anti-inflammatory effect during inflammation activation. However, the mechanisms involved in the anti-inflammatory effects of Bif-1 remain unclear.
Autophagy inhibitors increased the levels of inflammatory cytokines in LPS/ATP-induced J774A.1 cells
Autophagy involves the maintenance of normal cellular function and biological homeostasis through the breakdown and removal of damaged or unwanted cellular components (36). An increasing number of studies indicated that autophagy is also related to the regulation of inflammation (37,38).
It was shown that treatment with LPS/ATP significantly increased autophagy activity in J774A.1 cells, suggesting that LPS and ATP may influence cellular functions and physiological processes by regulating the autophagy pathway (Fig. 3A). The upregulation of p62 protein levels is shown in Fig. 3A. The LPS/ATP-induced inflammatory model is associated with NF-κB activation and activation of NF-κB can directly upregulate the level of p62 (30). This upregulation of p62 may enhance intracellular autophagy, which is triggered by factors such as oxidative stress and protein aggregation in the condition of inflammasome activation.
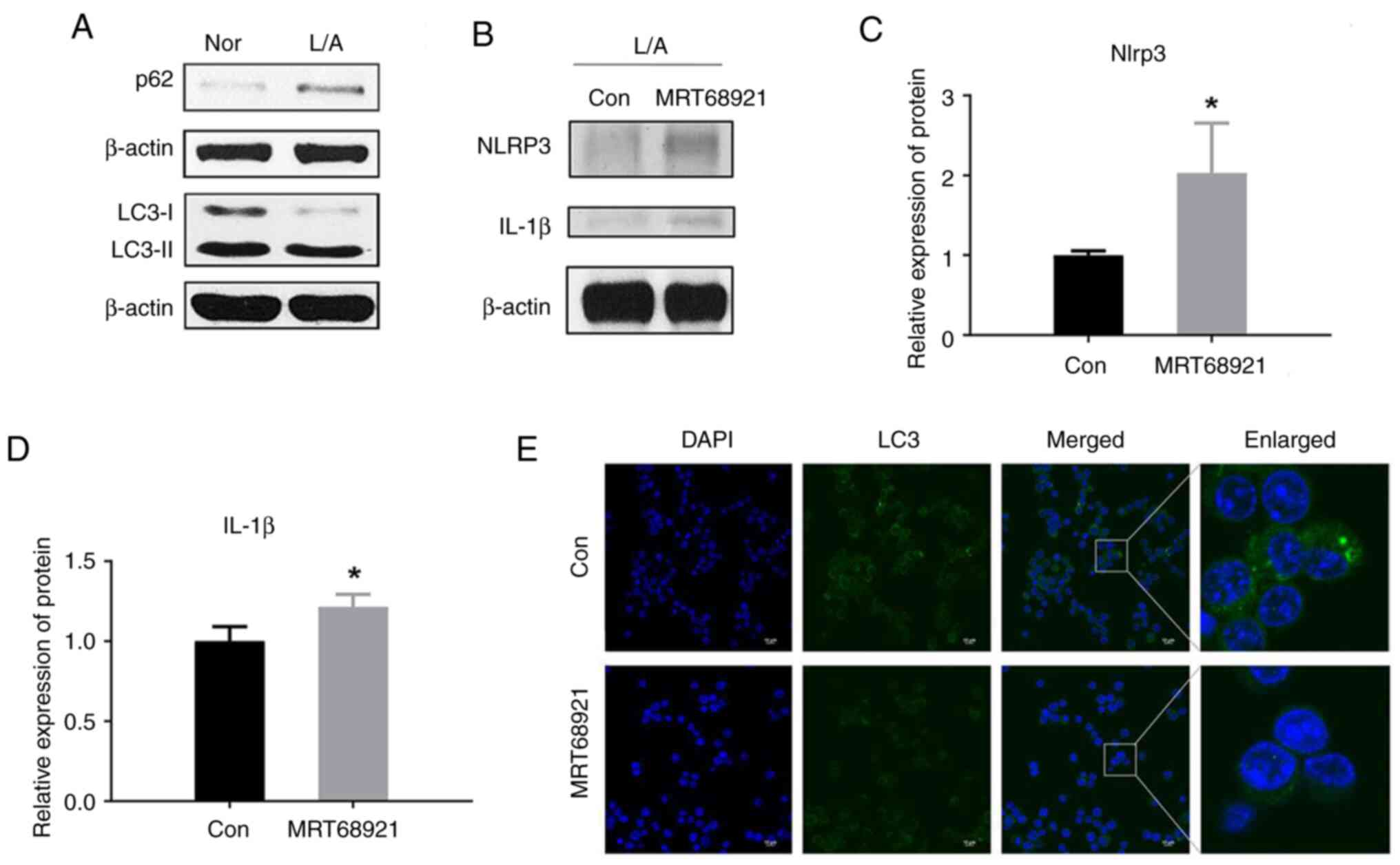
Meanwhile, the use of the autophagy inhibitor MRT68921, a ULK1 inhibitor, increased LPS/ATP-induced inflammatory cytokine levels (Fig. 3B-D). The serine/threonine-protein kinase ULK1 is a key molecule in the initiation of autophagy (39). Treatment with the autophagy inhibitor MRT68921 led to a reduction in the overall fluorescence of LC3 (Fig. 3E), indicating successful inhibition of autophagy. This implies that autophagy may play a protective role in suppressing inflammatory responses, and that autophagy inhibition may lead to increased inflammatory responses. These results provide insights about the relationship of autophagy with inflammatory regulation.
Based on these results, it was hypothesized that Bif-1 might exert an anti-inflammatory effect through a mechanism of autophagy regulation since Bif-1 is also an important autophagy regulation factor.
Use of si-Bif-1 significantly inhibits autophagy
Simultaneously, it was shown that the autophagy process was inhibited when the protein expression of Bif-1 was inhibited using si-Bif-1 (Fig. 4A and B). This suggested that Bif-1 plays an important role in the regulation of autophagy in inflammasome-activated cells, and its inhibition may lead to decreased autophagosome formation and degradation activity of inflammatory factors.
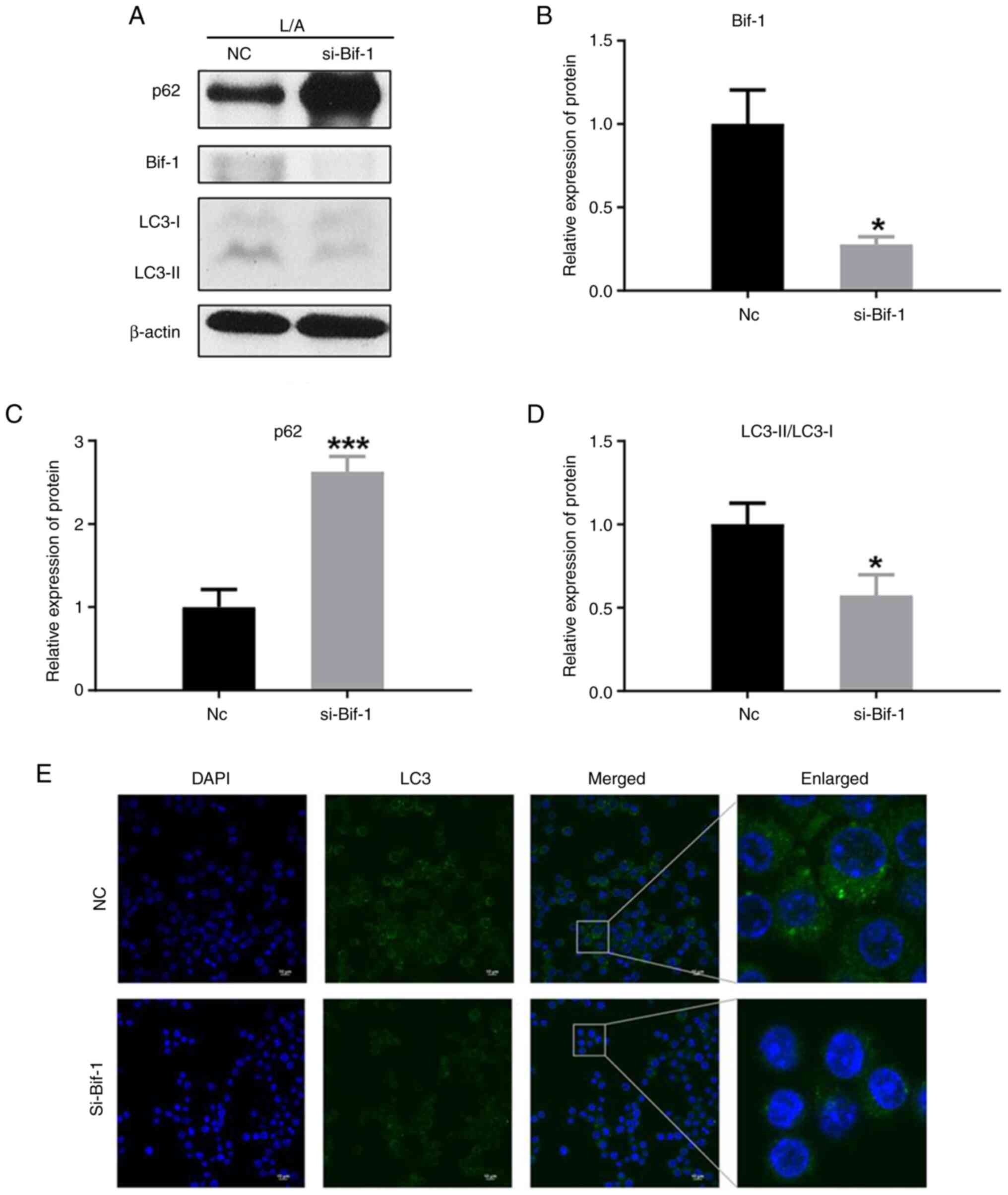
The level of p62 protein was increased after using si-Bif-1 (Fig. 4C). This was attributed to the fact that the decreased autophagic activity resulted in the inability of autophagosomes to efficiently degrade p62 protein, which led to its accumulation in cells. In the case of si-Bif-1 treatment, the action of inhibiting Bif-1 likely disrupts the degradation pathway of p62, thereby resulting in its upregulation.
It was also shown that the LC3-II/LC3-I ratio was decreased (Fig. 4D). LC3 is a key protein in the autophagy process, which undergoes phosphorylation and lipolysis to form LC3-I (40). Therefore, decreased autophagic activity leads to a decrease in the relative level of LC3-II. A decrease in the overall fluorescence of LC3 was observed when Bif-1 protein expression was inhibited using si-Bif-1 (Fig. 4E). This finding further supported the notion that Bif-1 plays a crucial role in the regulation of autophagy in inflammasome-activated cells, and its inhibition leads to the suppression of autophagy.
These results provided insights about the importance of Bif-1 in the regulation of autophagy and lay the foundation for in-depth exploration of the mechanism of Bif-1 in autophagy regulation.
NF-κB inhibitor significantly inhibited the increase of Bif-1 and autophagy in J774A.1 cells
There are two signaling pathways that activate NF-κB, and both cascades involve the activation of an IκB kinase (IKK) complex. MLN120B was used to block IKKβ phosphorylation to clarify the regulatory role of NF-κB signaling on Bif-1. Bif-1 protein levels were significantly downregulated in the MLN120B-treated group under LPS/ATP stimulation (Fig. 5A and B).
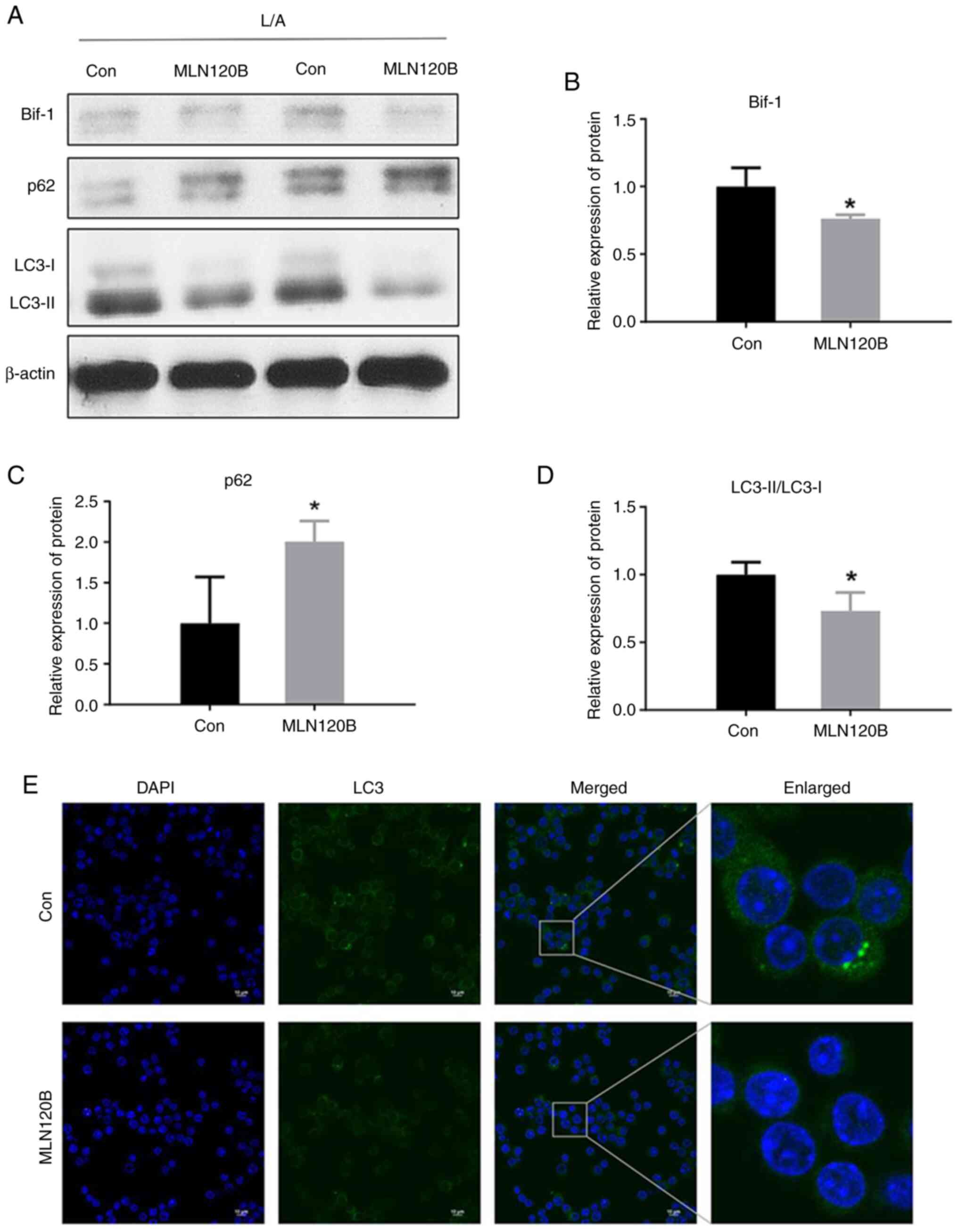
Meanwhile, the LC3-II protein level was downregulated in the MLN120B-treated group, and the p62 protein level was also affected (Fig. 5C and D). When treated with MLN120B, a decrease in the overall fluorescence of LC3 was observed, indicative of autophagy inhibition (Fig. 5E). These results further supported the importance of Bif-1 in the regulation of autophagy. Notably, total p62 protein level was increased after MLN120B treatment in the present study. The increased level of p62 is determined by numerous factors. NF-κB as a key factor can increase p62 and then enhance an early-stage autophagy and theoretically inhibition of NF-κB should decrease p62 (41). However, p62 can be degraded by a late-stage autophagy-lysosomal pathway and a significant inhibition of the pathway might result in the accumulation of intracellular p62 (42). Here, it seemed that p62 was mainly determined by MLN120B-induced autophagy inhibition instead of NF-κB inactivation. Further mechanisms should be investigated in the future. Despite this, these results provided a basis for further investigation of the relationship between NF-κB and Bif-1-mediated autophagy.
Discussion
The inflammatory response plays a key role in the development of a number of diseases. The regulation of the inflammatory response is important for the prevention and treatment of diseases (43). IL-1β is an important inflammatory mediator (44), the overproduction of which is closely associated with the development of a variety of inflammation-related diseases, and its inhibition has been shown to have therapeutic benefits in treating a range of severe but relatively rare inherited inflammatory diseases, such as cryopyrin-associated periodic syndromes and familial Mediterranean fever (45,46). IL-1β can activate the NF-κB signaling pathway, which in turn regulates the expression of a range of inflammation-related genes, including inflammatory mediators, cell adhesion molecules and inflammatory signaling molecules (47).
As an intracellular degradation mechanism, autophagic degradation involves a variety of pathophysiological processes, and it is generally considered to be a cytoprotective mechanism (48–50). Autophagy inhibits inflammasome formation and IL-1β production by degrading components of the NLRP3 inflammasome (37). Autophagosome is a key structure in the autophagy process, and its formation is the initiation stage of the autophagy process (51). Beclin-1 interacts with multiple cofactors such as Bif-1, Rubicon, Ambra1 and survivin, promoting the formation of the Beclin-1-Vps34-Vps15 complex (52). This complex triggers the cascade of autophagy reactions (53). Bif-1 plays an important regulatory role as a key protein in the autophagy process (19). In the present study, it was shown that the use of si-Bif-1 significantly inhibited autophagy. When Bif-1 expression is inhibited, autophagosome formation may be affected, thereby inhibiting autophagy. In addition, Bif-1 inhibition may also affect the expression of autophagy-related proteins, such as autophagy-related 5 and autophagy-related 9 (22). The functions of these proteins are closely related to the different steps of autophagy, and therefore the relationship between Bif-1 inhibition and autophagy needs to be investigated further.
In the current study, it was also shown that the expression of Bif-1 may be regulated by the NF-κB signaling pathway. It is hypothesized that there might be NF-κB binding sites in the promoter region of the Bif-1 gene, which allow NF-κB to directly or indirectly regulate the transcription of Bif-1. Alternatively, NF-κB signaling pathway may also be able to affect the expression of Bif-1 by regulating the activity of other transcription factors. Additional research is required to investigate these hypotheses. In the present study, a decrease of LC3 levels caused by MLN120B, an inhibitor of NF-κB signaling pathway, was observed, similar to what occurred with si-Bif-1. It seemed that both MLN120B and si-Bif-1 could inhibit autophagy. Since the NF-κB signaling pathway can affect Bif-1 and Bif-1 can affect autophagy, the results indicated that the NF-κB signaling pathway may affect autophagy through Bif-1. However, exact molecular mechanism should be investigated in the future.
Notably, in the present study, p62 levels were increased in a number of conditions, such as inflammasome activation induced by LPS/ATP, autophagy inhibition by si-Bif-1 and inhibition of the NF-κB signaling pathway by MLN120B. It seemed that p62 was affected by numerous separate factors as aforementioned. On one hand, as an autophagy adapter, increased levels of p62 facilitate the clearance of harmful protein aggregates and regulate oxidative stress, thereby maintaining cellular homeostasis. Furthermore, p62 can interact with inflammatory signaling molecules, modulating the activity of inflammatory signaling pathways (54). As aforementioned, the activation of the NF-κB signaling pathway in the condition of inflammation can upregulate p62 level (30). On the other hand, autophagic flux can affect p62 levels. Autophagic flux involves three steps: Nucleation, elongation and fusion (55). The initial nucleation of autophagic vesicles involves the class III phosphatidylinositol 3-kinase complex, comprising the kinase, Beclin-1 and UV radiation resistance-associated gene. Subsequently, LC3 undergoes lipidation, converting from the LC3 I to the LC3 II form. Finally, mature autophagosomes directly fuse with lysosomes or fuse with endosomes before being transported to lysosomes. Substrates within autolysosomes are then degraded and released into the cytoplasm. P62 localizes to autophagosomes through its interaction with LC3 and undergoes continuous degradation by the autophagy-lysosome system. Consequently, defective autophagy leads to the accumulation of p62 (56). Therefore, it seemed that early-stage autophagy activation and late-stage autophagy inhibition could increase p62 levels. It is clear that the use of MLN120B inhibited the NF-κB signaling pathway, however, the levels of p62 were likely still increased as a number of factors, other than NF-κB, contribute to the levels of p62.
The findings of the present study revealed the critical role of Bif-1 in the activation of the inflammasome. This finding is important for an in-depth understanding of the mechanisms of inflammatory response regulation. By revealing the key role of Bif-1 in the autophagy process, new insights were introduced into the understanding of the mechanisms of autophagy and inflammation. Further studies on the interactions of Bif-1 with other key proteins such as Beclin-1 and its regulatory mechanisms in inflammation and autophagy-related diseases will enable a deeper understanding of these complex biological processes. Further studies could explore the expression of Bif-1 in disease models and the relationship with inflammation- and autophagy-related signaling pathways to deepen the understanding of its mechanism of action.
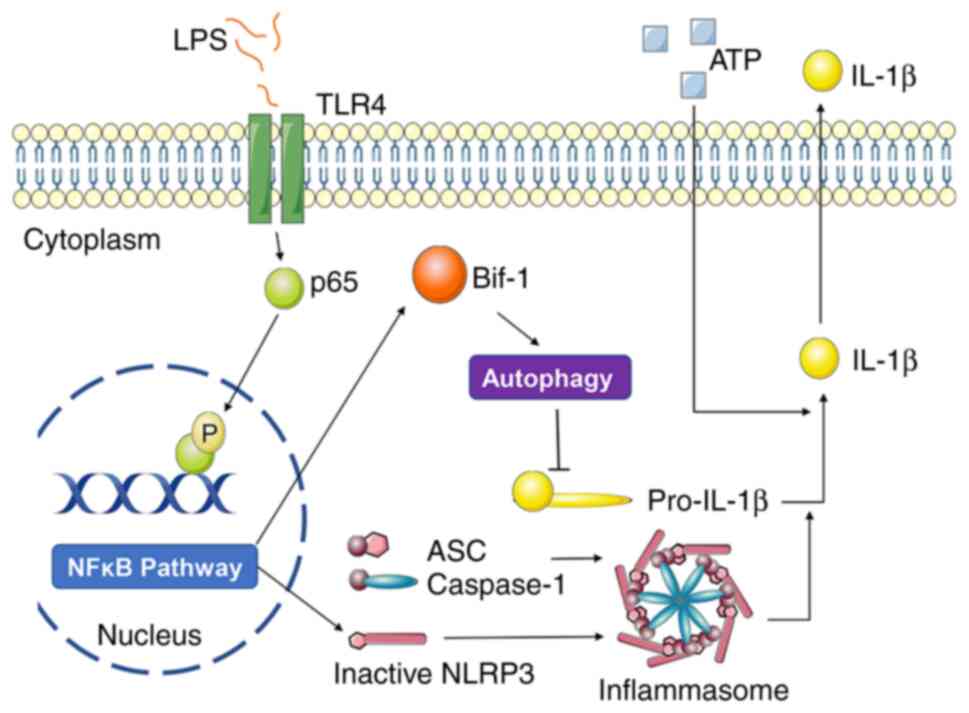
In conclusion, the findings of the present study revealed the mechanism used by Bif-1 to regulate inflammasome activation (Fig. 6), which provided new insights for elucidating the regulatory mechanism of inflammasome activation and provided the theoretical basis for the development of new drug targets.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by The Shenzhen Science and Technology Program (grant nos. JCYJ20190809160001751, JCYJ20210324111013036, RCJC20200714114433069 and JCYJ20200109142818589), and The People's Hospital of Baoan Shenzhen Scientific Research Project (grant no.BAYYZDXK001).
Availability of data and materials
The data generated in the present study may be requested from the corresponding author.
Authors' contributions
Data acquisition, processing and validation was performed by YZ, WS, LW and WX; the study investigation was carried out by YZ, WS, YN, HZ and LL; project administration was carried out by YZ, LW and WX; YZ, WS and WX wrote the original draft of the manuscript; YZ, LW and WX reviewed and edited the manuscript. YZ and WS confirm the authenticity of all the raw data. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
The animal study was reviewed and approved by The Bioethics Committee of Shenzhen International Graduate School, Tsinghua University [Shenzhen, China; approval no. (2020) No. 9].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
Medzhitov R: Origin and physiological roles of inflammation. Nature. 454:428–435. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Font MD, Thyagarajan B and Khanna AK: Sepsis and septic shock-basics of diagnosis, pathophysiology and clinical decision making. Med Clin North Am. 104:573–585. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Schroder K and Tschopp J: The inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Cornut M, Bourdonnay E and Henry T: Transcriptional regulation of inflammasomes. Int J Mol Sci. 21:80872020. View Article : Google Scholar : PubMed/NCBI | |
|
Roberti A, Chaffey LE and Greaves DR: NF-κB Signaling And Inflammation-Drug Repurposing To Treat Inflammatory Disorders? Biology (Basel). 11:3722022.PubMed/NCBI | |
|
Capece D, Verzella D, Flati I, Arboretto P, Cornice J and Franzoso G: NF-κB: Blending metabolism, immunity, and inflammation. Trends Immunol. 43:757–775. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Martinon F, Mayor A and Tschopp J: The inflammasomes: Guardians of the body. Annu Rev Immunol. 27:229–265. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Broz P and Dixit VM: Inflammasomes: Mechanism of assembly, regulation and signalling. Nat Rev Immunol. 16:407–420. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Lamkanfi M and Dixit VM: Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 28:137–161. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Swanson KV, Deng M and Ting JP: The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat Rev Immunol. 19:477–489. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Dinarello CA: Biologic basis for interleukin-1 in disease. Blood. 87:2095–2147. 1996. View Article : Google Scholar : PubMed/NCBI | |
|
Deretic V, Saitoh T and Akira S: Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 13:722–737. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Mizushima N, Levine B, Cuervo AM and Klionsky DJ: Autophagy fights disease through cellular self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Lopez-Castejon G: Control of the inflammasome by the ubiquitin system. FEBS J. 287:11–26. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A and Kehrl JH: Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 13:255–263. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE and Eissa NT: Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 27:135–144. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Kageyama S, Gudmundsson SR, Sou YS, Ichimura Y, Tamura N, Kazuno S, Ueno T, Miura Y, Noshiro D, Abe M, et al: p62/SQSTM1-droplet serves as a platform for autophagosome formation and anti-oxidative stress response. Nat Commun. 12:162021. View Article : Google Scholar : PubMed/NCBI | |
|
Heckmann BL, Boada-Romero E, Cunha LD, Magne J and Green DR: LC3-associated phagocytosis and inflammation. J Mol Biol. 429:3561–3576. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mulé JJ, et al: Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 9:1142–1151. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, Singh S and Wang HG: Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J Biol Chem. 276:20559–20565. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Pierrat B, Simonen M, Cueto M, Mestan J, Ferrigno P and Heim J: SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics. 71:222–234. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Takahashi Y, Meyerkord CL and Wang HG: BARgaining membranes for autophagosome formation: Regulation of autophagy and tumorigenesis by Bif-1/Endophilin B1. Autophagy. 4:121–124. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Xu L, Wang Z, He SY, Zhang SF, Luo HJ, Zhou K, Li XF, Qiu SP and Cao KY: Bax-interacting factor-1 inhibits cell proliferation and promotes apoptosis in prostate cancer cells. Oncol Rep. 36:3513–3521. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Frangež Ž, Fernández-Marrero Y, Stojkov D, Seyed Jafari SM, Hunger RE, Djonov V, Riether C and Simon HU: BIF-1 inhibits both mitochondrial and glycolytic ATP production: Its downregulation promotes melanoma growth. Oncogene. 39:4944–4955. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Schlauder SM, Calder KB, Khalil FK, Passmore L, Mathew RA and Morgan MB: Bif-1 and Bax expression in cutaneous Merkel cell carcinoma. J Cutan Pathol. 36:21–25. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Wang DB, Uo T, Kinoshita C, Sopher BL, Lee RJ, Murphy SP, Kinoshita Y, Garden GA, Wang HG and Morrison RS: Bax interacting factor-1 promotes survival and mitochondrial elongation in neurons. J Neurosci. 34:2674–2683. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Karbowski M, Jeong SY and Youle RJ: Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 166:1027–1039. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Niu Y, Zhang Y, Zhang W, Lu J, Chen Y, Hao W, Zhou J, Wang L and Xie W: Canagliflozin ameliorates NLRP3 inflammasome-mediated inflammation through inhibiting NF-κB signaling and upregulating Bif-1. Front Pharmacol. 13:8205412022. View Article : Google Scholar : PubMed/NCBI | |
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Xu C, Wang W, Zhong J, Lei F, Xu N, Zhang Y and Xie W: Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem Pharmacol. 152:45–59. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
He Y, Hara H and Núñez G: Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 41:1012–1021. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Meyers AK, Wang Z, Han W, Zhao Q, Zabalawi M, Duan L, Liu J, Zhang Q, Manne RK, Lorenzo F, et al: Pyruvate dehydrogenase kinase supports macrophage NLRP3 inflammasome activation during acute inflammation. Cell Rep. 42:1119412023. View Article : Google Scholar : PubMed/NCBI | |
|
Hayden MS and Ghosh S: NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 26:203–234. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Hsu CG, Li W, Sowden M, Chávez CL and Berk BC: Pnpt1 mediates NLRP3 inflammasome activation by MAVS and metabolic reprogramming in macrophages. Cell Mol Immunol. 20:131–142. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Hseu YC, Tseng YF, Pandey S, Shrestha S, Lin KY, Lin CW, Lee CC, Huang ST and Yang HL: Coenzyme Q0 inhibits NLRP3 inflammasome activation through mitophagy induction in LPS/ATP-stimulated macrophages. Oxid Med Cell Longev. 2022:42662142022. View Article : Google Scholar : PubMed/NCBI | |
|
Ma Y, Galluzzi L, Zitvogel L and Kroemer G: Autophagy and cellular immune responses. Immunity. 39:211–227. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Deretic V: Autophagy in inflammation, infection, and immunometabolism. Immunity. 54:437–453. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Qiu P, Liu Y and Zhang J: Review: The role and mechanisms of macrophage autophagy in sepsis. Inflammation. 42:6–19. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Petherick KJ, Conway OJ, Mpamhanga C, Osborne SA, Kamal A, Saxty B and Ganley IG: Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 290:11376–11383. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Criollo A, Senovilla L, Authier H, Maiuri MC, Morselli E, Vitale I, Kepp O, Tasdemir E, Galluzzi L, Shen S, et al: The IKK complex contributes to the induction of autophagy. EMBO J. 29:619–631. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 171:603–614. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Freeman TL and Swartz TH: Targeting the NLRP3 inflammasome in severe COVID-19. Front Immunol. 11:15182020. View Article : Google Scholar : PubMed/NCBI | |
|
Dinarello CA: Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 281:8–27. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Menu P and Vince JE: The NLRP3 inflammasome in health and disease: The good, the bad and the ugly. Clin Exp Immunol. 166:1–15. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, Brik R, McCann L, Kasapcopur O, Rutkowska-Sak L, et al: Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 367:2396–2406. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Lawrence T: The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 1:a0016512009. View Article : Google Scholar : PubMed/NCBI | |
|
Lei Y and Klionsky DJ: Transcriptional regulation of autophagy and its implications in human disease. Cell Death Differ. 30:1416–1429. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Münz C: Chapter Five-autophagy in immunity. Progress in Molecular Biology and Translational Science. Vol 172. Martinez AB and Galluzzi L: Academic Press; pp. 67–85. 2020, View Article : Google Scholar : PubMed/NCBI | |
|
Gan T, Qu S, Zhang H and Zhou XJ: Modulation of the immunity and inflammation by autophagy. MedComm (2020). 4:e3112023.PubMed/NCBI | |
|
Zhao YG, Codogno P and Zhang H: Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat Rev Mol Cell Biol. 22:733–750. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Martinez J, Malireddi RKS, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J, et al: Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 17:893–906. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Toton E, Lisiak N, Sawicka P and Rybczynska M: Beclin-1 and its role as a target for anticancer therapy. J Physiol Pharmacol. 65:459–467. 2014.PubMed/NCBI | |
|
Moscat J and Diaz-Meco MT: p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 137:1001–1004. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Yu L, Chen Y and Tooze SA: Autophagy pathway: Cellular and molecular mechanisms. Autophagy. 14:207–215. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Liu Y, Liu HQ, Xiao JY, Ma KT, Wang XQ, Shen HJ and Luo JD: Autophagy is involved in the protective effect of endophilin A2 on H2O2-induced apoptosis in H9C2 cardiomyocytes. Biochem Biophys Res Commun. 499:299–306. 2018. View Article : Google Scholar : PubMed/NCBI |