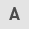DNAJA1‑knockout alleviates heat stroke‑induced endothelial barrier disruption via improving thermal tolerance and suppressing the MLCK‑MLC signaling pathway
- Authors:
- Published online on: March 26, 2024 https://doi.org/10.3892/mmr.2024.13211
- Article Number: 87
-
Copyright: © Li et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Heat stroke (HS) is a life-threatening condition clinically characterized by extreme hyperthermia (>40.5°C), central nervous system dysfunction, systematic inflammatory response syndrome and multiorgan dysfunction syndrome (1). According to its cause and degree of susceptibility within the population, HS can be categorized as either classic or exertional, although both are associated with high morbidity and mortality rates, and warrant attention and research (2). It has been reported that episodes of extreme heat waves will increase in the future, meaning that the incidence of HS is likely to increase further (3). Furthermore, a previous study from Kyoto indicated that the estimated number of patients with HS transported by ambulance is predicted to be >3× greater among people aged 0–64 years and 6× greater among people aged ≥65 years (4). Therefore, it is necessary to explore the medical intervention methods against HS.
The endothelium, a barrier formed by squamous endothelial cells, is a highly dynamic layer that mainly lines the blood vessels and is responsible for maintaining the stability of microvascular permeability (5). Accumulating evidence has suggested that the disruption of the normal vascular endothelial barrier plays a key role during the pathophysiological processes of HS (1,6). The vascular endothelium exhibits discontinuity in HS, which manifests as increased intercellular gaps and extensive endothelial cell apoptosis, ultimately leading to compromised structural-barrier integrity (6). The increase in vascular endothelial permeability is the main mechanism underlying capillary leakage, which results in severe coagulation problems (7). Protection of the vascular endothelial barrier has resulted in beneficial effects on HS such as inhibition of coagulopathy and reduction of the risk of vascular leakage, although the specific mechanisms underlying this have not been revealed (7,8). The present study conducted genome-wide CRISPR-Cas9 screening research (Fig. S1) to explore the specific genes responsible for resisting HS and enhanced heat tolerance in the early stage using human umbilical vein endothelial cells (HUVECs) (9,10).
DNAJA1 is a protein-coding gene encoding DnaJ heat shock protein (HSP)40 family member A1, which is often considered to act as a HSP70 co-chaperone, and facilitate protein folding and trafficking and prevent aggregation (11,12). Traditionally, HSPs serve a crucial role in the cellular response to heat stress by maintaining protein homeostasis and preventing protein denaturation (13). However, few studies have investigated the effects of DNAJA1-KO on HS, and its protective effects on HS go against the conventional theories surrounding HSPs. Of note, a previous study revealed that HSP90 inhibitors prevent lipopolysaccharide (LPS)-induced endothelial barrier dysfunction by suppressing RhoA signaling (14). As HUVECs were used to identify heat tolerance-related genes, the present study aimed to investigate the protective effects and mechanisms of DNAJA1-KO on HS-induced endothelial barrier disruption. Specifically, it was demonstrated that DNAJA1 may be a novel potential therapeutic target for regulating the damage of vascular endothelial barrier during HS.
Materials and methods
Cell culture and HS protocol
Immortalized HUVECs (cat. no. 1101HUM-PUMC000437; Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences) were cultured in DMEM (Wuhan Servicebio Technology Co., Ltd.) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin at 37°C in 5% CO2. The control cells were incubated at 37°C, while HS was induced in the experimental cells via incubation at 43°C for 4 h, before returning to 37°C for further research (12). The cell culture was refreshed every 2 days to remove non-adherent cells. EDTA-trypsin (Gibco; Thermo Fisher Scientific, Inc.) was used to passage the cells when the cells reached 80% confluence. The HUVECs were divided into Control (CON) and DNAJA1-KO groups according to whether or not they were subjected to CRISPR/Cas9 gene KO; these two groups were further divided into CON, CON + HS, DNAJA1-KO and KO + HS groups according to whether they underwent HS treatment.
Construction of a stable cell line
CRISPR/Cas9 technology was used to construct a gene KO cell line. The single guide RNA (sgRNA; forward, 5′-ACCGCAGCTCGTTGAAGCACTGTG-3′) targeting the transcription start site of the DNAJA1 gene exon 1 (Gene ID: NM_001314039; 5′-CAGCTCGTTGAAGCACTGTG-3′) and the control sgRNA (5′-ACGGAGGCTAAGCGTCGCAA-3′) were designed using VectorNTI (version, 11.5.1; Invitrogen; Thermo Fisher Scientific, Inc.) and induced into the pLenti-U6-spgRNA v2.0-CMV-Puro-P2A-3×FLAG-spCas9-WPRE plasmid vector [OBiO Technology (Shanghai) Corp., Ltd.]. Subsequently, the two plasmids were transduced into HUVECs according to the recommended protocol of OBiO. The sgRNA for knockout of DNAJA1 was used in KO groups and the control sg RNA was used in CON groups. The efficiency of DNAJA1 knockout was confirmed by western blotting.
Cell viability assays
To measure cell viability, HUVECs were analyzed using a Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Inc.) assay and a lactate dehydrogenase (LDH) Cytotoxicity Assay Kit-WST (LDH; Dojindo Laboratories, Inc.). Cells were seeded in a 96-well plate at a density of 1×105/ml and incubated at 37°C overnight. Subsequently, cells were treated according to the group, mixed with CCK-8 solution and incubated for 2 h. According to the manufacturer's instructions, the absorbances of CCK-8 and LDH were measured using a microplate reader (ND-171; Omega Bio-Tek, Inc.) at wavelengths of 450 and 490 nm, respectively.
Flow cytometric analysis
Apoptosis was assessed using an Annexin V-FITC Apoptosis Detection Kit [cat. no. AT101; Multi Sciences (Lianke) Biotech Co., Ltd.] according to the manufacturer's instructions. Briefly, HUVECs (~1×106) were collected, washed with pre-cooled phosphate-buffered saline (PBS), and resuspended in binding buffer containing 5 µl Annexin V-FITC and 10 µl PI. The samples were then incubated at room temperature for 5–15 min in the dark, before running on a flow cytometer (FACSCalibur; BD Biosciences). The results were analyzed using Cell Quest Pro software (version 6.0; BD Biosciences). The apoptosis rate was calculated as: Apoptosis rate (%)=(apoptotic cell number/total cell number) ×100.
Endothelial monolayer permeability assays
Transendothelial electrical resistance (TEER) and FITC-Dextran permeability assays were conducted to evaluate the barrier function of HUVECs. Briefly, cells were seeded on Transwell filters (0.4-µm-pore, 6.5-mm-diameter, 24-well plates; cat. no. 356008; Corning, Inc.) coated with fibronectin which had been incubated for 24 h at 37°C in 5% CO2 at a density of 1×106 cells/ml and were incubated at 37°C in 5% CO2. The cells seeded on Transwell filters were also cultured in DMEM (Wuhan Servicebio Technology Co., Ltd.) with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin. Each Transwell insert was placed in a well with 200 µl medium (DMEM with 10% FBS) in the upper chamber and 1 ml medium (DMEM with 10% FBS) in the lower chamber. After reaching 100% confluence, an EVOM2 Epithelial Voltohmmeter (World Precision Instruments) was used to detect the TEER values of each group. The TEER (in Ω/cm2) was calculated using the following equation: (resistance of sample well-resistance of corresponding blank well) ×0.33, where 0.33 is the area (in cm2) of the insert membrane. The TEER was monitored daily and was considered to form the integrated endothelial monolayer when the values remained stable after increasing. As aforementioned, the integrated endothelial monolayer samples were cultured in an incubator at 43°C for 4 h to simulate HS injury, while the corresponding control samples were cultured in a normal incubator at 37°C. After HS treatment, the TEER was detected in each group. The measurements were recorded in triplicate and are reported as the TEER values (mean + standard error) comparing four groups. Furthermore, based on the integrated endothelial monolayer model using Transwell filters, the cell medium was washed and refreshed with HEPES medium (Beyotime Institute of Biotechnology), before adding FITC-Dextran (Invitrogen; Thermo Fisher Scientific, Inc.) to the upper chamber. The plates were incubated at 37°C in 5% CO2 for 4 h and the samples were then aspirated from the bottom chamber to a solid black plate and fluorescence intensity (excitation/emission wavelength, 490/520 nm) was read using a fluorometer (ND-171; Omega Bio-Tek, Inc.).
Western blot analysis
After corresponding treatments, HUVEC monolayers grown in 6-well plates (Corning, Inc.) were lysed on ice using RIPA lysis buffer (Beyotime Institute of Biotechnology) with a protease inhibitor cocktail and phosphatase inhibitor cocktail (Roche Diagnostics GmbH), before centrifuging cell lysates at 15,000 × g at 4°C for 10 min. Subsequently, the protein concentration in the supernatant was measured using the bicinchoninic acid (BCA) protein assay kit (Beyotime Institute of Biotechnology), before detecting the protein expression by western blotting. Protein samples were mixed with loading buffer, denatured and separated on 4–12 and 4–20% SurePAGE™ gels (GenScript) with 30 µg protein per lane. Proteins were transferred to PVDF membranes using an eBlot™ L1 Fast Wet Transfer System (GenScript). The blotted wet membrane was blocked for 2 h at room temperature with blocking buffer (QuickBlock Western Blocking Buffer; cat. no. P0252; Beyotime Institute of Biotechnology). Next, the membranes were incubated overnight with primary antibodies at 4°C, followed by incubation with secondary antibodies for 2 h at 37°C. The Omni-ECLTM Femto Light Chemiluminescence Kit (cat. no. SQ201; Epizyme, Inc.; Ipsen Biopharmaceuticals, Inc.) was used to detect luminescence signals on the membrane, and the Amersham Imager 600 (Cytiva) was used to image the blots. The protein expression was quantified using ImageJ software (version 1.52e; National Institutes of Health). The following primary antibodies were used overnight at 4°C: DNAJA1 (Proteintech Group, Inc.; cat. no. 11713-1-AP; 1:1,000; rabbit), GAPDH (Beyotime Institute of Biotechnology; cat. no. AF0006; 1:1,000; mouse), cleaved-caspase-3 p17 (Cell Signaling Technology, Inc.; cat. no. 9661; 1:1,000; rabbit), caspase-3 (ABclonal Biotech Co., Ltd.; cat. no. A22869; 1:500; rabbit), Bcl-2 (Proteintech Group, Inc.; cat. no. 68103-1-Ig; 1:8,000; mouse), Bax (Proteintech Group, Inc.; cat. no. 60267-1-Ig; 1:8,000; mouse), zonula occludens-1 (ZO-1; Cell Signaling Technology, Inc.; cat. no. 13663; 1:1,000, rabbit), junctional adhesion molecule A (JAM-A; Cell Signaling Technology, Inc.; cat. no. 82196; 1:1,000; rabbit), claudin-5 (Abcam; cat. no. ab131259; 1:1,000; rabbit), occludin (Invitrogen; Thermo Fisher Scientific, Inc.; cat. no. 33-1500; 1:1,000; mouse), phosphorylated (p)-myosin light-chain kinase (MLCK; Abcam; cat. no. ab200809; 1:1,000; rabbit), MLCK (Santa Cruz Biotechnology, Inc.; cat. no. sc-365352; 1:1,000; mouse), p-MLC (Cell Signaling Technology, Inc.; cat. no. 3675S; 1:1,000; mouse) and MLC (Cell Signaling Technology, Inc.; cat. no. 3672S; 1:1,000; rabbit). The following secondary antibodies were used: 1:8,000, Goat Anti-Mouse IgG (H+L) HRP (1:8,000, cat. no. AB0102, Shanghai Abways Biotechnology Co., Ltd.); and Goat Anti-Rabbit IgG (H+L) HRP (1:8,000, cat. no. AB0101, Shanghai Abways Biotechnology Co., Ltd.).
Immunofluorescence analysis
Cells were washed three times with PBS and fixed in 4% paraformaldehyde and 2% Triton X-100 at room temperature for 10 min, before incubating with ZO-1 (Cell Signaling Technology, Inc.; cat. no. 13663; 1:100, rabbit) and JAM-A (Cell Signaling Technology, Inc.; cat. no. 82196; 1:200, rabbit) antibodies overnight at 4°C. Following incubation, the samples were incubated with Alexa Fluor 488-labeled Goat Anti-Rabbit IgG(H+L) secondary antibodies (1:500, cat. no. A0423, Beyotime Institute of Biotechnology) and TRITC phalloidin for labeling F-actin (Beijing Solarbio Science & Technology Co., Ltd.) for 2 h at room temperature. After washing, the cells were stained with DAPI for 5 min at room temperature (cat. no. C1006, Beyotime Institute of Biotechnology) and visualized using an inverted fluorescence microscope (IX71; Olympus Corporation).
Proteomic analysis
Cell samples were disrupted in lysis buffer (8M urea, 1% SDS) and the concentrations were determined using a BCA kit according to the manufacturer's protocol. Protein digestion was performed according to the standard procedure. Subsequently, the peptides were subjected to vacuum drying, followed by resuspension with a solution containing 2% acetonitrile and 0.1% trifluoroacetic acid. The samples were then desalted using Sep-Pak and underwent vacuum drying. The peptide concentrations were determined using a peptide quantification kit (cat. no. 23275; Thermo Fisher Scientific, Inc.). Loading buffer was added to each tube to prepare samples for mass spectrometry analysis and the concentration of each sample was 0.5 µg/µl. To enhance the proteomic depth, the samples were fractionated through high pH reversed-phase separation. The peptides were reconstituted using a loading buffer consisting of ammonium hydroxide solution containing 2% acetonitrile and adjusted to pH 10. Separation of the peptides was performed by Shanghai Majorbio Bio-pharm Technology Co., Ltd., using high pH reversed-phase liquid chromatography (LC) (RPLC; Acquity Ultra Performance LC; Waters Corporation). The gradient elution was performed on a high pH RPLC column (Acquity Ultra Performance Liquid Chromatography Ethylene Bridged Hybrid C18 Column, 1.7 µm, 2.1×150 mm, Waters Corporation) at 200 µl/min with the gradient increased for 66 min (phase B, ammonium hydroxide solution containing 80% acetonitrile, pH 10; phase A: ammonium hydroxide solution containing 2% acetonitrile, pH 10). A total of 10 fractions were obtained per sample by pooling 20 fractions from each. LC-tandem mass spectrometry (MS/MS) (EASY-nLC 1200, Thermo Fisher Scientific, Inc.) analysis was performed by Shanghai Majorbio Bio-pharm Technology Co., Ltd. The sample was loaded onto a C18-reversed phase column (75 µm × 25 cm; Thermo Fisher Scientific, Inc.) in buffer A (2% acetonitrile and 0.1% formic acid) and separated with a linear gradient of buffer B (80% acetonitrile and 0.1% formic acid) at a flow rate of 300 nl/min. An electrospray voltage of 1.8 kV against the inlet of the mass spectrometer was used. The nitrogen temperature was 22°C (room temperature in the mass spectrometry laboratory) and the positive ionization mode was used. A Q Exactive mass spectrometer (Thermo Fisher Scientific, Inc) was operated in the data-dependent mode, to switch automatically between MS and MS/MS acquisition. Survey full-scan MS spectra (m/z 350-1,300) were acquired with a mass resolution of 70K, followed by twenty sequential high energy collisional dissociation MS/MS scans with a resolution of 17.5K. In all cases, one microscan was recorded using dynamic exclusion of 18 sec. MS/MS spectra were searched using Proteome Discoverer Software 2.2 software (Thermo Fisher Scientific, Inc.) against the Homo sapiens database from UniProt website (https://www.uniprot.org/proteomes/UP000005640). The identification of parent proteins was accomplished using the peptide mass with the highest score, which corresponded to the predicted value in the database. The parameters employed for protein searching were established as follows: Tryptic digestion with a maximum of two missed cleavages, fixed modification of carbamidomethylation of cysteines, and variable modification of oxidation of methionines and protein N-terminal acetylation. Validation of peptide spectral matches was executed through the assessment of q-values at a 1% false discovery rate. In the present study, a total of 4,394 proteins expressed were identified as belonging to the proteome of HUVECs. The thresholds of the fold change (>1.2 or <0.83) and P<0.05 were used to identify differentially expressed proteins (DEPs). Annotation of all identified proteins was performed using Gene Ontology (GO) term (http://www.blast2go.com/b2ghome; http://geneontology.org/) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (http://www.genome.jp/kegg) enrichment analyses. The data were analyzed using the free online Majorbio Choud platform (cloud.majorbio.com) (15). Raw proteomics data are available on ProteomeXchange (https://www.proteomexchange.org/) with accession number PXD043512.
Animals
HS protocols and sample collectionMale C57BL/6J mice (age, 7–8 weeks; weight, 18–22 g) were purchased from Shanghai SIPPR-Bk Laboratory Animal Co., Ltd. Standard pellet rat chow and distilled tap water were provided ad libitum. A total of 12 mice (CON (Wild type mice), n=3; KO (DNAJA1−/− mice), n=3; CON (Wild type mice)CON + HS, n=3; and KO + HS (DNAJA1−/− mice), n=3) were used for the experiments. All mice were raised in the Specific Pathogen Free Animal Experiment Center of the Naval Medical University (Shanghai, China) and were acclimated to the animal facility conditions (22±1°C; humidity, 50±5%; 12-h light/dark cycle) for 1 week prior to the experiment. All experimental procedures were approved by The Institutional Animal Ethics Committee of the Naval Medical University (approval no. NMUMREC-2021-002) according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (16). All mice in this experiment were anesthetized with isoflurane (induction, 3–5%; maintenance, 1–2%) using a gas anesthesia machine (KW-MZJ; Nanjing Calvin Biotechnology Co., Ltd.). Humane endpoints of mice unable to live normally due to severe pain and discomfort caused by the experimental procedures were included in the experimental design, however no mice reached these endpoints in the present study. The animals were placed in the chamber and perfused with CO2 at a rate of 30% volume/min until the animals were not moving, not breathing and their pupils were dilated. Subsequently, the CO2 was turned off, and the animals were observed for 5 min to confirm that the animals had been sacrificed. DNAJA1-KO mice with C57BL/6 genetic background were generated and purchased by Cyagen Biosciences, Inc. All mice were genotyped by PCR analysis. In brief, the PCR genotyping identification protocol for gene knockout mice involved tail sampling (a 2 mm section of the end of the mouse tail was and stored at −80°C), DNA extraction was performed using a Servicebio Genomic DNA Kit (cat. no. G3633, Wuhan Servicebio Technology Co., Ltd.), PCR amplification was performed using 2× Fast Pfus PCR Master Mix (cat. no. G3305, Wuhan Servicebio Technology Co., Ltd.), gel electrophoresis was performed using a 2% agarose gel and visualized using. SerRed nucleic acid dye (G3606, Wuhan Servicebio Technology Co., Ltd.) and a Gel Imaging System (WD-9413B; Beijing Liuyi Biotechnology Co., Ltd.). Briefly, PCR amplification was performed as follows, pre-denaturation at 98°C for 2 min, then 30 cycles of denaturation at 98°C for 20 sec, annealing at 55°C for 20 sec and extension at 72°C for 10 sec before a final extension at 72°C for 5 min. Two primer sequences were designed to identify the knockout mice. The primer sequences targeting DNAJA1−/− mice genotyping (designed by GemPharmatech Co., Ltd.) were as follows: Primer 1 forward (F), 5′-CGGATATTCAGAAAGGATTGGGAC-3′ and reverse (R), 5′-ACCTTGAACTCACTGTGAAGCCAA-3′ (product for wild type, 7354 bp; product for KO, 325 bp). Primer 2 F, 5′-TCAACTTTCTGAGTACCAGGTCC-3′ and R, 5′-CAAAGCCTCATCCCTGAAGAGA-3′ (product for wild type, 349 bp; product for KO: 0 bp). The banding patterns expected were as follows: Wild type with Primer 1, a single wild type band; wild type with Primer 2, a single wild type band; heterozygote with Primer 1, a wild type band and a KO band; heterozygote with Primer 2, a wild type band; homozygote (DNAJA1−/−): with Primer 1, a single KO band; and homozygote (DNAJA1−/−) with Primer 2 reaction, no product.
The improved HS protocol was based on our previously reported study (17). Before HS induction, a temperature-monitoring capsule (SV223 capsule thermometer; Shenzhen Flamingo Technology Co., Ltd.) with real-time temperature monitoring function was implanted in the experimental mice to obtain the core body temperature (Tc) at 5-min intervals. An artificial climate chamber (LTH-575N-01; Shanghai Drawell Instrument Co., Ltd.), where ambient temperature was controlled at 40±1°C and relative humidity at 60±5% to create an environment with high temperature and humidity, was used to induce HS onset. The timepoint of stable hyperthermia occurrence (Tc >42.7°C) was taken as the primary criterion of HS onset in mice. The use of humane endpoints in animal research was mainly based on the criteria for HS by monitoring the core body temperature using temperature-monitoring capsules mentioned above (8). If the mice met the criteria of hyperthermia occurrence (Tc >42.7°C), they were sampled and euthanized 3 h after the onset of HS. Control mice not subjected to heat stress were sampled and euthanized at the same time as experimental mice (3 h after the onset of HS).
Blood sample examination and histological examination
Blood samples were collected from the abdominal aorta and separated by centrifugation at 1,000 × g for 10 min at 4°C, then sub-packed and stored at −80°C for later examination. Serum biochemical indices of alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea (BU), creatine kinase (CK) and creatinine (CREA) were examined using an automatic biochemical analyzer (HITACHI 7080 automated analyzer; Hitachi, Ltd.). Serum von Willebrand factor (VWF, cat. no. D721155, Sangon Biotech Co., Ltd.), thrombomodulin (TM, cat. no. D721144, Sangon Biotech Co., Ltd.) and endothelin-1 (ET-1, cat. no. D721180, Sangon Biotech Co., Ltd.) were respectively detected using corresponding ELISA kits according to the manufacturer's instructions. Lung tissue samples were treated according to the protocol described in our previous study (17). Briefly, the organ samples were fixed in 4% paraformaldehyde at room temperature for >24 h, dehydrated, embedded, sectioned to a thickness of 4–6 µm using a rotary microtome and stained with hematoxylin and eosin (H&E) for 3–5 min and 15 sec at room temperature, respectively. All the H&E-stained slides were examined using a light microscope (Leica DM2000; Leica Microsystems GmbH) and scanned using a Panoramic MIDI Slide scanner (3DHISTECH, Ltd.). Paraffin-embedded sections of the lung samples were also used for immunofluorescence staining. Sections were incubated with xylene at room temperature for 15 min, twice, and then rehydrated in a descending alcohol series. The sections were then immersed in sodium citrate antigen retrieval solution (pH 6.0) and heated to 99°C in the pressure cooker and held for 3 min before air cooling. Sections were incubated with 3% bovine serum albumin for 30 min to block non-specific binding at room temperature. Sections were incubated with primary antibodies against VE-cadherin (1:500; cat. no. AF8319; rabbit; Beyotime Institute of Biotechnology) overnight at 4°C. Sections were rinsed with water and then incubated with Cy3-conjugated Goat Anti-Rabbit IgG secondary antibodies (1:500; cat. no. GB21301500; rabbit; Wuhan Servicebio Technology Co., Ltd.) at room temperature for 50 min in dark condition. Sections were washed with PBS and then counterstained with DAPI solution at room temperature for 10 min, in the dark. Spontaneous fluorescence was quenched by incubation with Antifluorescence Quenching Mounting Media (G1401, Wuhan Servicebio Technology Co., Ltd.) for 5 min at room temperature. Sections were imaged using a Nikon Eclipse TI-SR fluorescent microscope (Nikon Corporation). CY3 glows red by excitation wavelength 510–560 nm and emission wavelength 590 nm. DAPI glows blue by UV excitation wavelength 330–380 nm and emission wavelength 420 nm. The image acquisition system (Nikon DS-U3; Nikon Corporation) and microscope settings were maintained throughout the process.
Statistical analysis
Data are presented as the mean ± standard error of mean. The sample size for the cell viability and endothelial monolayer permeability assays was six replicates per group. The sample size for the flow cytometric analysis, western blot analysis, proteomic analysis and animal experiments was three replicates per group. Unpaired Student's t-test was used to compare two groups. One-way ANOVA with Bonferroni's post-hoc test was used for comparisons among multiple groups. The statistical values were calculated using GraphPad Prism (version 9.4.0, GraphPad Software; Dotmatics) with P<0.05 considered to indicate a statistically significant difference.
Results
DNAJA1-KO improves the thermal tolerance of HUVECs following HS
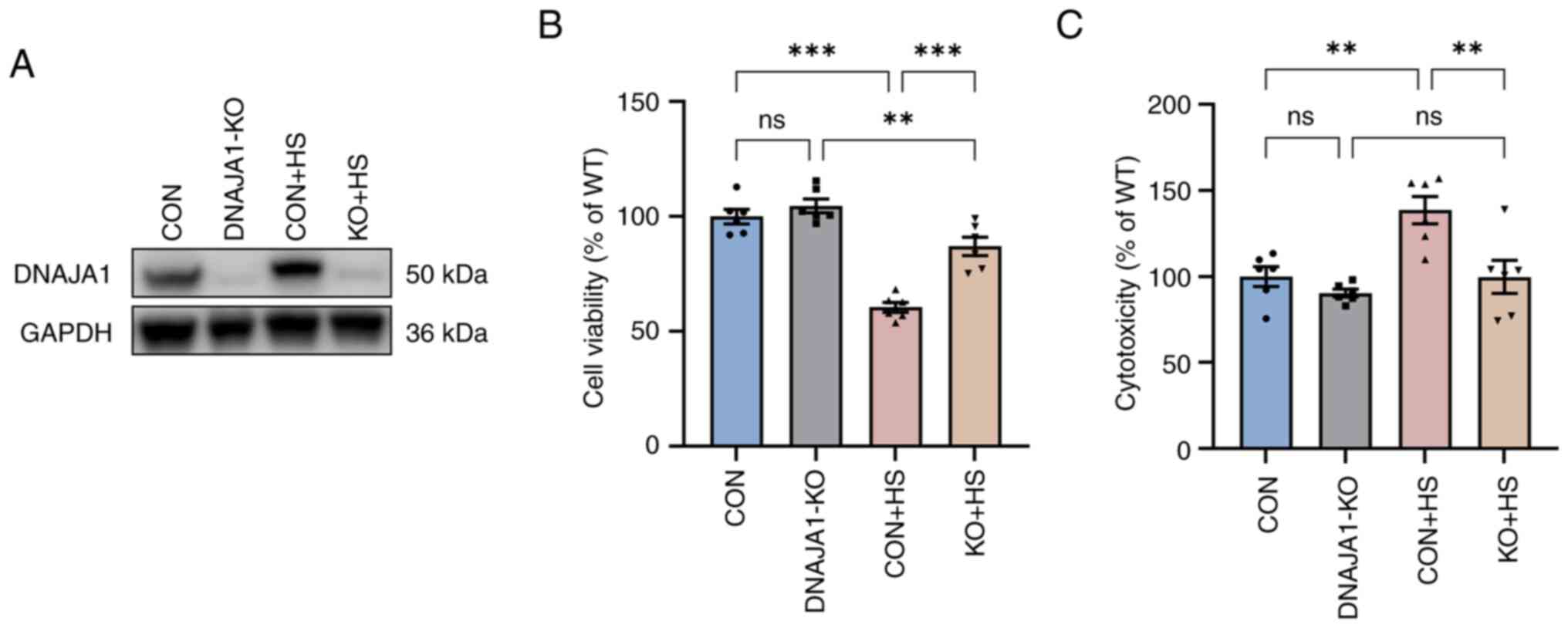
The stable DNAJA1-KO cell line was constructed successfully, according to the results of western blotting (Fig. 1A). The CCK-8 assay demonstrated that DNAJA1-KO had no significant effect on cell viability, whereas it significantly improved viability compared with the CON + HS cells incubated at 43°C for 4 h (Fig. 1B). A previous study reported that HS exerts a cytotoxic effect on HUVECs (18,19). DNAJA1-KO cells could markedly withstand HS-induced cytotoxic effects, as indicated by the concentration of LDH detected by the LDH release assay (Fig. 1C). Furthermore, HS-induced endothelial apoptosis has been widely confirmed in the early stages of experimental heat stress (20). Next, the early apoptosis rate of HS-treated cells was determined by flow cytometry. The results demonstrated that the apoptosis rate of HS-treated CON cells was significantly increased compared with that of untreated CON cells, whereas only a few DNAJA1-KO cells were apoptotic (Fig. 2A and C). In parallel, western blot analysis confirmed that DNAJA1-KO could alleviate HS-induced apoptosis, as demonstrated by the apparent reduction of cleaved caspase-3 by KO in HS cells (Fig. 2B). Notably, the classic apoptosis-inducing protein Bax showed no difference in expression, whereas the classic anti-apoptotic protein Bcl-2 was upregulated in DNAJA1-KO cells (21). Taken together, these data demonstrated that DNAJA1-KO significantly improved the thermal tolerance of HUVECs, as indicated by the results of cell viability and apoptosis assays.
DNAJA1-KO alleviates HS-induced endothelial barrier dysfunction
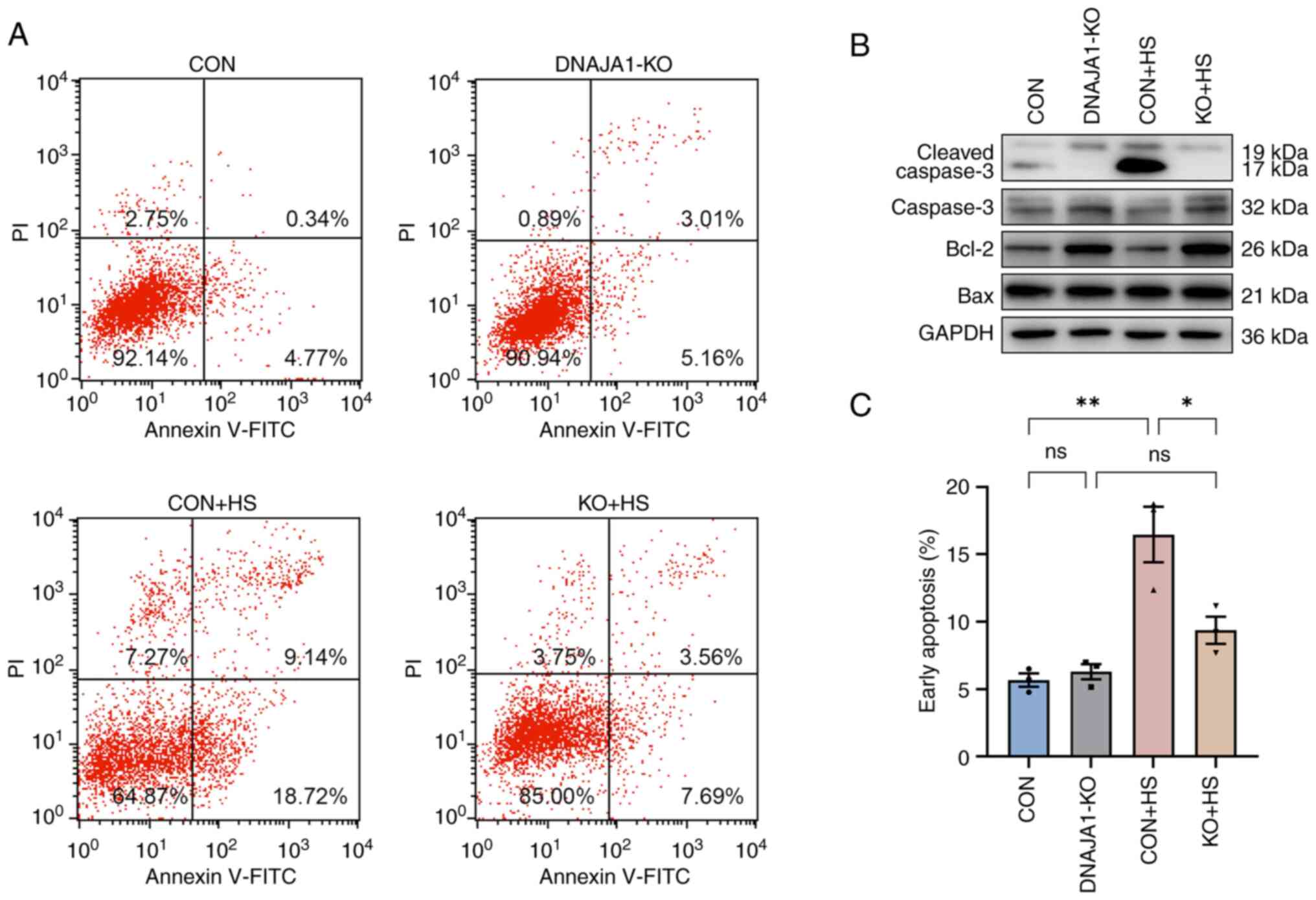
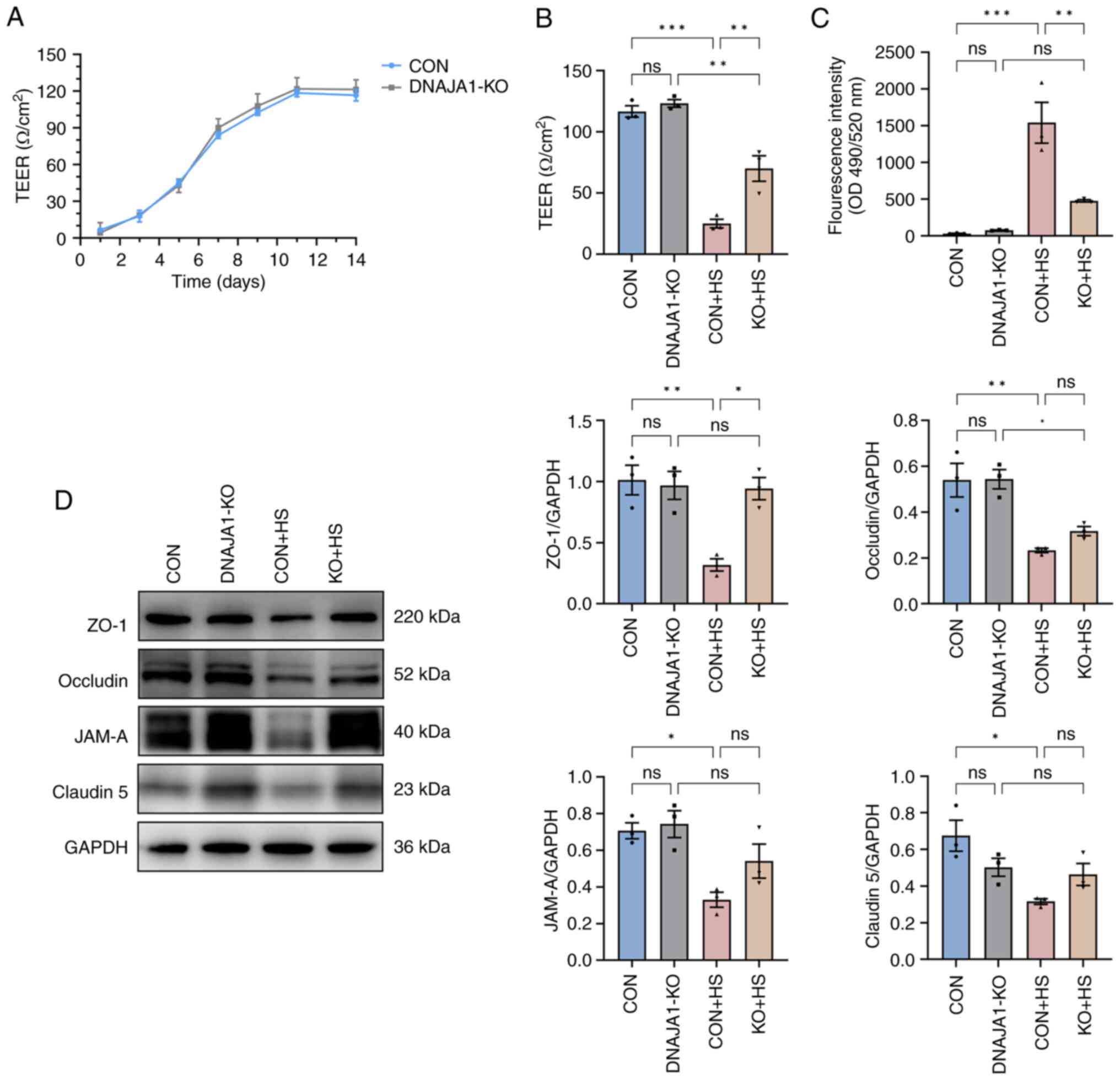
After the cells were seeded on Transwell filters, three wells were sampled from CON cell-derived monolayers and DNAJA1-KO cell-derived monolayers to monitor the integrity of the cells. Once the TEER values became stable, endothelial monolayer permeability assays were conducted to determine the effects of DNAJA1-KO. Based on the monitored TEER, the endothelial monolayer met the experimental requirements and was generally >100 Ω/cm2 (Fig. 3A). The integrated endothelial monolayer was incubated at 43°C for 4 h to simulate HS injury, while the corresponding control filters were cultured in a normal incubator. The results demonstrated that TEER of the CON + HS group decreased, while more FITC-Dextran penetrated the damaged monolayer to the bottom chamber of the Transwell. By contrast, the KO + HS group maintained partial integrity of the endothelial monolayer (Fig. 3B and C). As endothelial barrier function could be protected by DNAJA1-KO, investigation into the condition of the structure that maintained the barrier was prompted. Numerous studies have proposed that the endothelial barrier shares a number of features with tight junctions (TJs) in epithelial cells (22,23). Therefore, the expression of TJ proteins in each group was assessed by western blotting to estimate the key structure of the barrier. Under heat stress for 4 h, the expression of TJ proteins, including ZO-1, occludin, JAM-A and claudin-5, decreased significantly in CON + HS cells compared with that in the CON cells (Fig. 3D). The expression of these representative TJ proteins was normalized to GAPDH and statistical analysis was performed. Although the expression of occludin, JAM-A and claudin-5 in KO + HS cells was enhanced compared with that in CON + HS cells, these differences were not statistically significant. Of note, the expression of ZO-1 was effectively upregulated in KO + HS with statistical significance, which strongly demonstrated that the endothelial barrier of DNAJA1-KO cells showed greater heat resistance ability against HS.
DNAJA1-KO protects against HS-induced damage of the cytoskeleton and TJs
Although the expression of TJ proteins was maintained in DNAJA1-KO cells, the arrangement of the TJ proteins into a particular formation was required for the TJs to act as a functional barrier (22). It has also been reported that both the downregulation of TJ protein expression and the alterations of subcellular localization of TJ proteins contribute to barrier dysfunction (23). ZO-1 and JAM-A were examined by immunofluorescence assay to evaluate the structural integrity of the TJs. As shown in Fig. 4, in the CON and DNAJA1-KO cell monolayers without HS, ZO-1 staining exhibited an integrated and continuous structure with belt-like lines. However, this continuous structure was difficult to observe in CON + HS cells, and the overall fluorescence intensity also decreased compared with that in untreated CON cell monolayers. Correspondingly, a clear structure of ZO-1 staining could be observed in KO + HS, with a slight decrease in fluorescence intensity compared with KO (Fig. 4A). Similar results were obtained for JAM-A, which indicated that the disrupted structure of JAM-A IN CON + HS was protected by DNAJA1-KO (Fig. 4B). Meanwhile, TRITC phalloidin was used to investigate the changes in F-actin in HUVECs under HS. F-actin is a key cytoskeletal component, which is dynamic in the context of cell-cell junctions and has been reported to be rearranged under HS (8,24,25). Similar to the findings of previous studies (8), the cortical actin rim became incomplete, the inter-cell gap widened, and stress fibers thickened and reorganized in CON + HS cells subjected to HS at 43°C, all of which was alleviated in KO + HS cells (Fig. 4A and B).
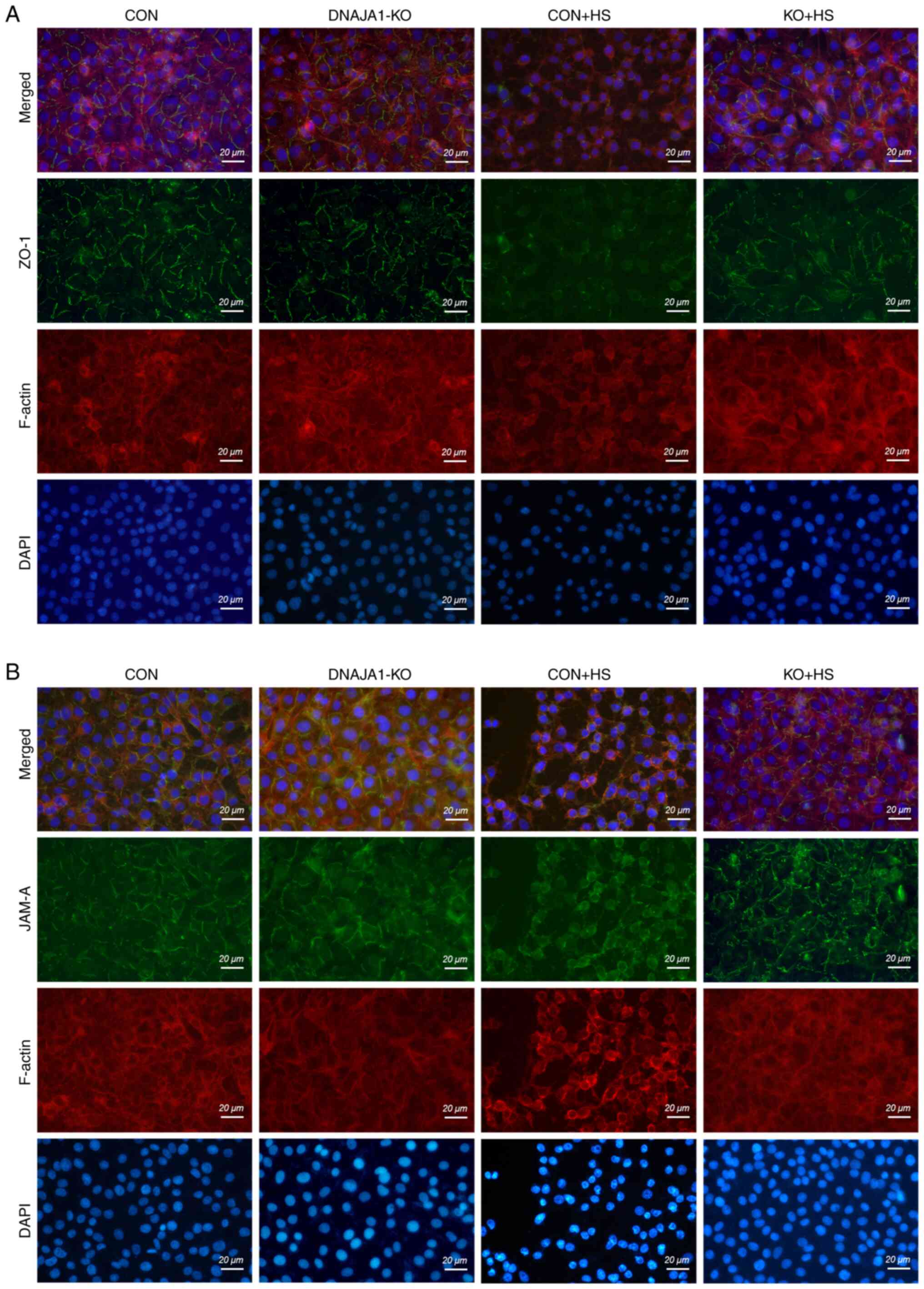
Proteomics analysis and identification of DEPs in HUVECs
A proteomics experiment using a label-free technique was performed to analyze and identify the DEPs of the four groups of HUVECs. A total of 4,394 expressed proteins were identified as belonging to the proteome of HUVECs in the present study. The distribution of these proteins in the four groups is shown in a Venn diagram (Fig. 5A). These proteins were analyzed statistically with thresholds of fold change (>1.2 or <0.83) and P<0.05. As a result, 2,499 proteins were identified as DEPs and were plotted in statistical histograms and volcano plots (Fig. 5B and C). Furthermore, Venn diagrams (Fig. 5D) were employed to analyze the distribution of upregulated proteins in CON + HS compared with CON cells (CON + HS vs. CON up), downregulated proteins in KO + HS compared with CON + HS cells (KO + HS vs. CON + HS down), downregulated proteins in CON + HS compared with CON cells (CON + HS vs. CON down) and upregulated proteins in KO + HS compared with CON + HS cells (KO + HS vs. CON + HS up). In the present study, the protein expression differences in CON vs. CON + HS and KO + HS vs. CON + HS were mainly focused on. As a result, 102 DEPs were found to be upregulated in CON + HS compared with CON and downregulated in KO + HS compared with CON + HS. These proteins, activated by HS and inhibited by KO, could be described as ‘bad’ proteins. Correspondingly, 98 DEPs were downregulated in CON + HS compared with CON, and upregulated in KO + HS compared with CON + HS. These proteins, activated by HS and inhibited by KO, could be described as ‘good’ proteins. Analysis of these screened proteins could help to clarify the mechanisms underlying the protective effects of DNAJA1-KO.
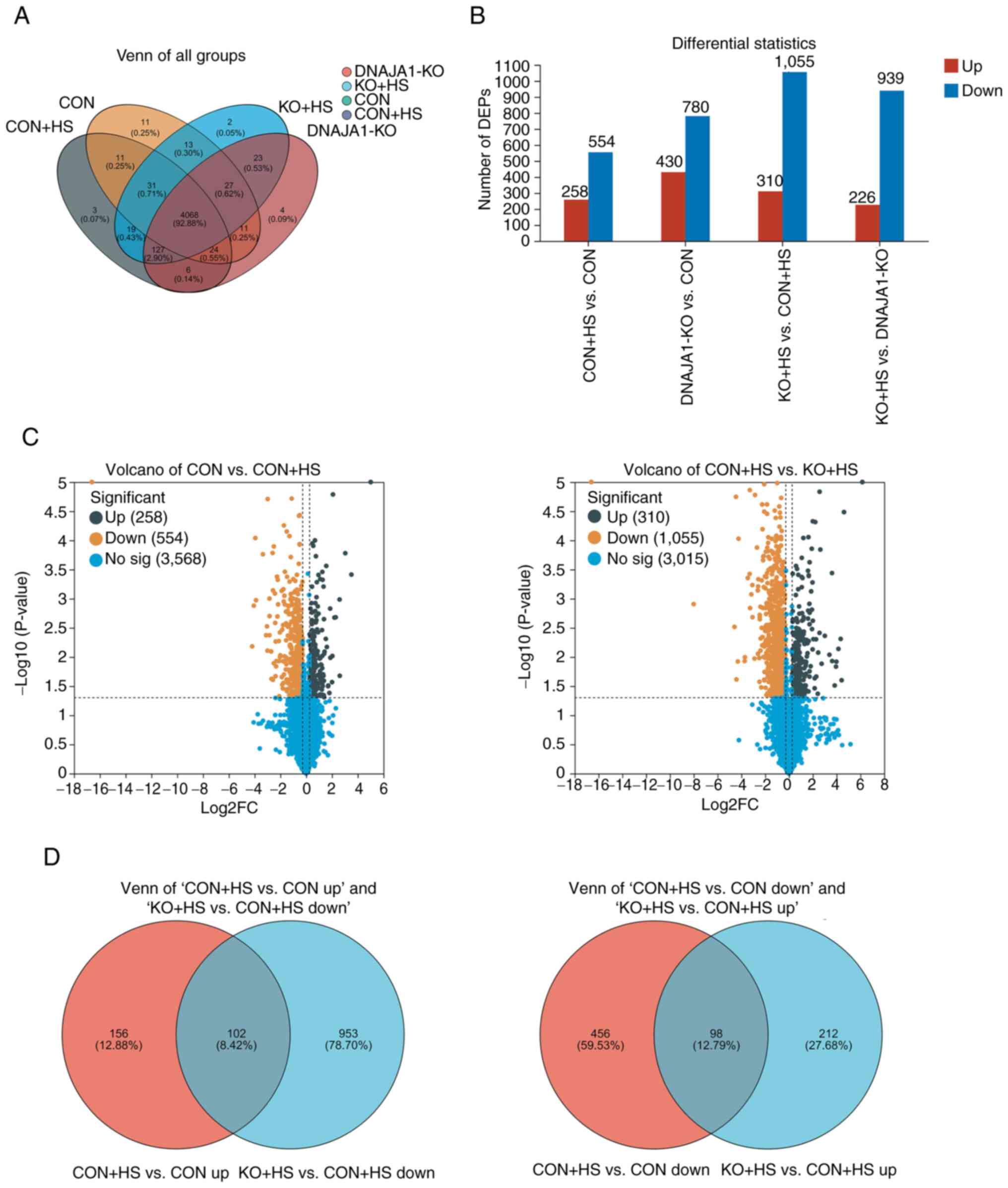
Analysis of interaction networks for DEPs
KEGG and GO enrichment analyses were performed to improve the understanding of the functional basis and biological pathways of the aforementioned screened DEPs. KEGG enrichment analysis of the 102 DEPs revealed that 10 pathways were significantly enriched (Fig. 6A), among which, the ‘calcium signaling pathway’ and vascular smooth muscle contraction were highly relevant to the present study. Therefore, these two pathways were further analyzed and the associated proteins framed in blue (Fig. S1B-E). Coincidently, the two pathways were similar and shared various DEPs with parallel regulation, including MLCK and calmodulin. GO enrichment analysis of the 102 DEPs demonstrated that the enriched pathways were associated with a number of protein-folding processes (Fig. 6B). KEGG enrichment analysis of the 98 DEPs revealed that 13 pathways were significantly enriched (Fig. 6D). GO enrichment analysis of the 98 DEPs revealed that the enriched pathways were associated with numerous DNA replication-related processes (Fig. 6E). Enriched complement and coagulation cascades may be related to endothelial injury-induced coagulopathy in HS. As the 102 ‘bad’ DEPs could provide novel potential pathways, a heat map was used to classify the DEP expression pattern between ‘CON + HS vs. CON up’ and ‘KO + HS vs. CON + HS down’ (Fig. 6C). Hierarchical clustering of the 102 DEPs showed distinguished expression patterns between the four groups. Particularly, proteins framed in red echoed the results of the aforementioned results of the present study and were demonstrated to be significant.
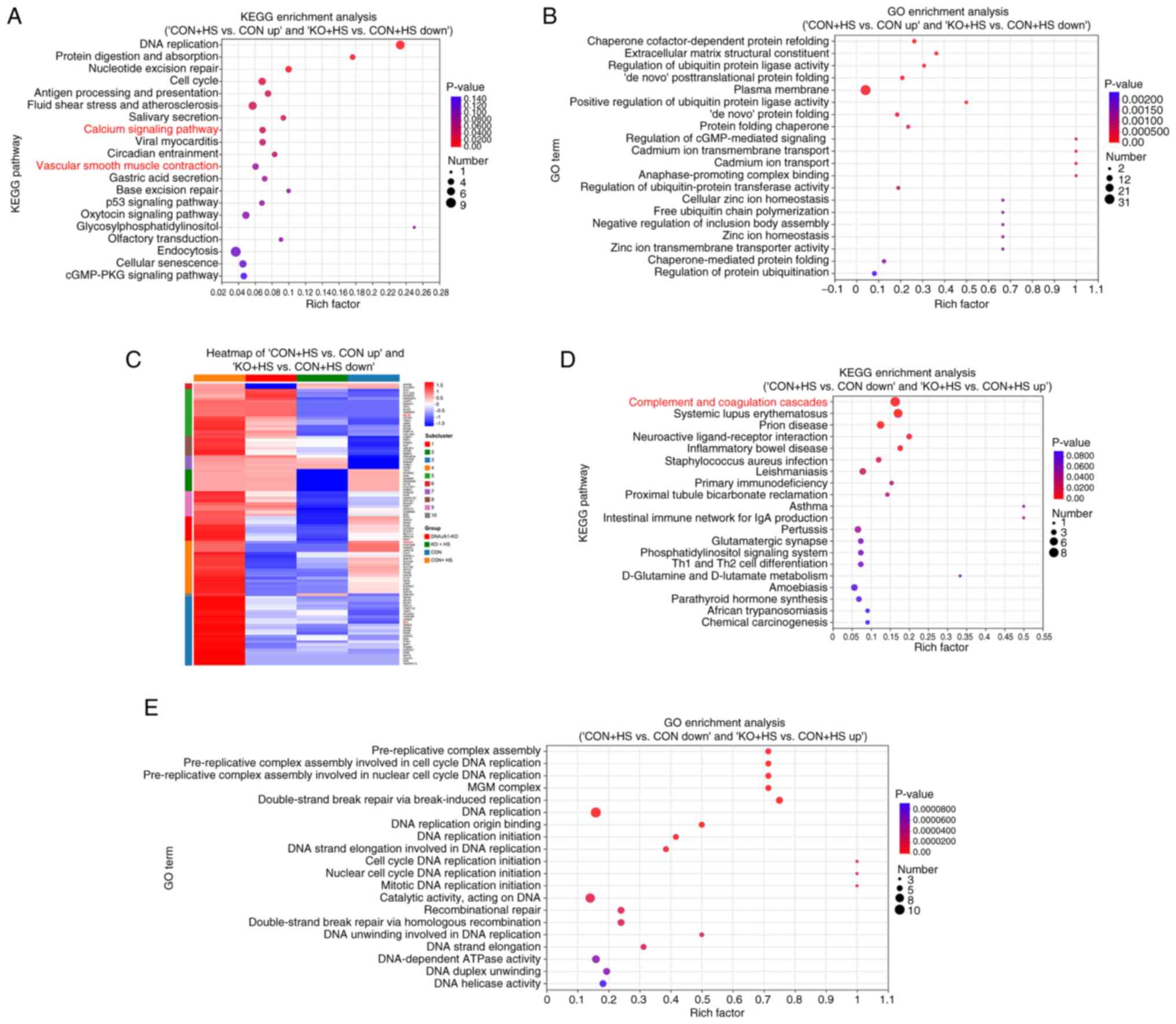
DNAJA1-KO on HS is regulated by the MLCK-MLC signaling pathway
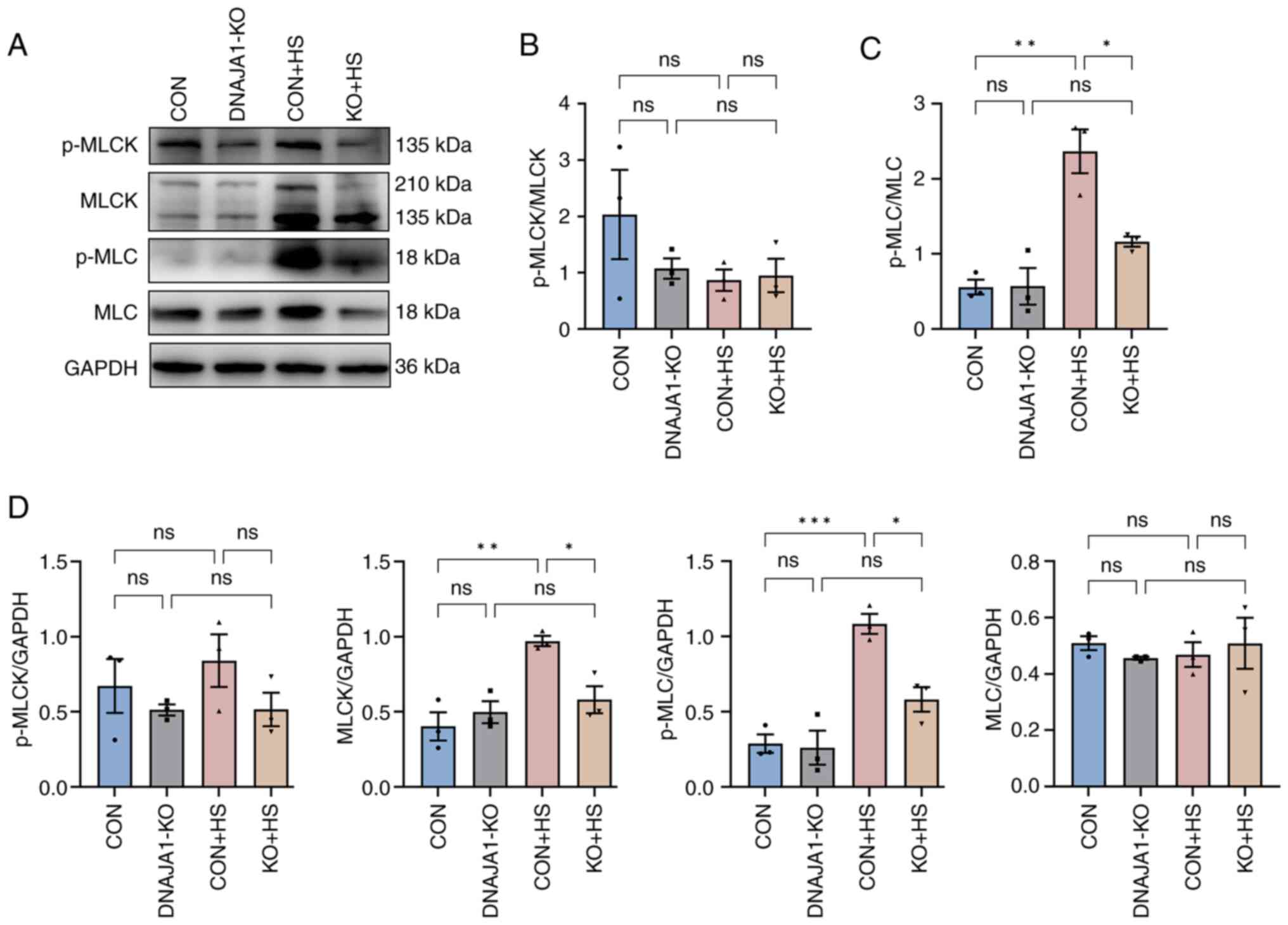
It is widely recognized that MLCK plays a key role in the regulation of TJs, and the associated endothelial and epithelial barrier (26). Moreover, pathways that were found to be enriched by proteomic analysis confirmed that MLCK had a negative or pro-destructive role in the protection of DNAJA1-KO. To further investigate the MLCK-related signaling pathway, MLCK, MLC, p-MLCK and p-MLC were examined by western blotting and semi-quantified relative to GAPDH (Fig. 7A). Consistent with the proteomics results, MLCK expression was significantly elevated in CON + HS cells and inhibited by DNAJA1-KO in KO + HS cells. The expression levels of p-MLC were accordingly increased in CON + HS cells and reduced in KO + HS cells, whereas no statistical difference was found in p-MLCK and MLC between the groups (Fig. 7D). The ratio of p-MLCK to total MLCK (Fig. 7B) and that of p-MLC to total MLC (Fig. 7C) were semi-quantified to further verify the mechanism. The ratio of p-MLCK to total MLCK was not statistically significant, whereas the ratio of p-MLC to total MLC was significantly higher in CON + HS cells than that in both CON and KO + HS groups, indicating that the activation of p-MLC may be associated with MLCK but not with unchanged MLC. These results suggested that DNAJA1-KO could alleviate HS-induced endothelial barrier disruption by downregulating the elevated MLCK expression and reducing p-MLC expression.
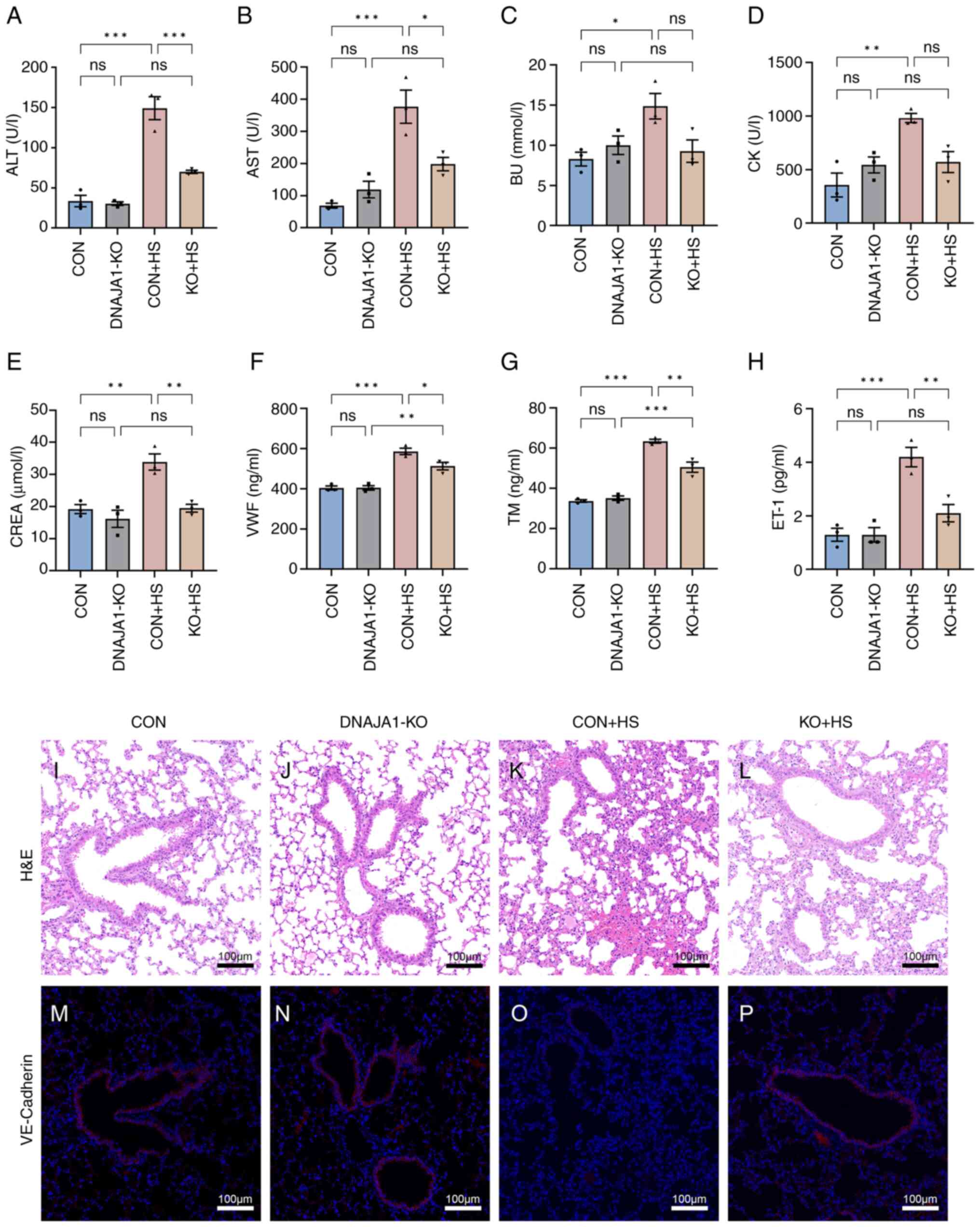
DNAJA1-KO attenuates multiple-organ injury and endothelial barrier dysfunction in HS mice
All KO mice were genotyped by PCR analysis (Fig. S1F). Multiple-organ injury in mice of each group was evaluated by detecting biochemical markers that represent the related function of each organ. Serum biochemical markers of liver function (ALT and AST), kidney function (BU and CREA), and skeletal and/or cardiac muscle function (CK) were determined to evaluate organ injury. As shown in Fig. 8A-E, the marked organ injury of CON + HS mice was markedly inhibited by DNAJA1-KO. Among the biochemical markers, although BU and CK were not significantly decreased, a downward trend was apparent. In addition, VWF, TM and ET-1 were selected to evaluate the degree of vascular endothelial injury (Fig. 8F-H). Significant high levels of serum vascular endothelial injury biomarkers were detected in CON + HS, while the levels of these three biomarkers were relatively low in KO + HS. Consistent with the serum biomarkers, lung, as an organ rich in vascular endothelial cells, showed alveolar wall thickening, narrowed alveolae, numerous monocytes scattered with granulocytes and severe congestion in vessels from CON + HS mice (Fig. 8K). Typical lung injury was attenuated by DNAJA1-KO as shown in Fig. 8L. VE-cadherin, which represents vascular endothelial function, had a markedly lowered expression in the lungs of mice subjected to HS induction (Fig. 8O). The expression of VE-cadherin was notably higher in the KO + HS group (Fig. 8P) compared with the CON + HS group, which indicated that DNAJA1-KO could counteract the HS-induced lung vascular endothelial barrier dysfunction.
Discussion
The endothelium, formed by squamous endothelial cells, is a highly dynamic layer that lines the heart, blood vessels, lymph vessels and serous cavities of the body (26). It is involved in numerous physiological functions, including filtrating fluid, forming a barrier, maintaining vascular tissues, regulating immune function and other organ-specific functions (27). Endothelial responses are damaged by exposure to heat stress and play a key role in the pathophysiology of HS (7). Heat stress not only damages the endothelium directly in the acute phase, but also indirectly disrupts endothelial structures and functions through the inflammatory immune response (28). Numerous studies have found that endothelial cells can be damaged by HS a high temperature of 43°C (29–31). To identify the specific gene associated with thermal tolerance, genome-wide CRISPR-Cas9 screen research was conducted on HUVECs (10). The increased cell viability and decreased concentration of LDH release preliminarily proved that DNAJA1-KO improved the thermal tolerance of HUVECs. A number of researchers consider that cells become apoptotic immediately after HS (18,19,29,32). The present study showed that the rate of early apoptotic cells was significantly decreased in KO + HS cells. At this point, it was proposed that the ameliorative effects of DNAJA1-KO on cell damage or death could be interpreted as an improvement in thermal tolerance. As aforementioned, DNAJA1 belongs to the HSP40 family and is essential for the heat shock response. A previous study found that HSP90 protein inhibitors may help to protect and restore endothelial barrier function following LPS treatment via regulating myosin phosphatase target subunit 1 (14,33). Notably, another study suggested that the HSP70 protein can play a protective role in sepsis by maintaining endothelial permeability via p38 signaling (34). Studies have indicated that HSP proteins have a huge number of families and are associated with various biological effects (13). To the best of our knowledge, DNAJA1, as a member of the HSP40 family, has not been previously investigated in terms of its effects on the endothelial barrier. The notable protective effects of DNAJA1-KO on HS led to further investigations into whether it could help sustain endothelial barrier integrity. Therefore, a confluent monolayer of HUVECs was successfully constructed by monitoring the TEER according to previously reported methods (35,36). It has been reported that a decrease in TEER inversely reflects a change in endothelial monolayer permeability (37). Endothelial monolayer permeability assays showed that the barrier function disruption of HS could be inhibited by DNAJA1-KO. The maintenance of endothelial junction integrity is governed by proteins that establish adhesion and TJs, thereby facilitating intercellular communication in a dynamic manner. Endothelial cell TJs share numerous features with TJs in epithelial cells and have been explored widely. Several representative TJ proteins were downregulated by HS and upregulated by DNAJA1-KO. Staining of ZO-1 and JAM-A further confirmed these barrier protective effects. Moreover, the endothelial cytoskeleton plays a critical role in barrier function and regulation. The hyperpermeability of the endothelial monolayer is attributed to the destabilization of actin microfilaments or disassembly of microtubules (38). Growing evidence has indicated that HSP proteins participate in the organization of the cytoskeleton in various cell lines such as the HaCaT and HeLa cell lines (39–42) It was also found that the structure of F-actin was damaged by HS and mitigated by DNAJA1-KO, which could sustain the cytoskeleton structure. To date, the protective effects of DNAJA1-KO on HS-induced endothelial barrier disruption have only been classified at a basic level.
Proteomic analysis was conducted to determine the potential pathways responsible for the specific underlying mechanism of DNAJA1-KO in HS. It was hypothesized that proteins activated by HS and inhibited by DNAJA1-KO could play a pro-damage role, while those inhibited by HS and activated by DNAJA1-KO could play an anti-damage role. As a result, 102 ‘pro-damage’ proteins and 98 ‘anti-damage’ proteins were identified, while the subsequent enrichment analysis provided potential pathways to verify these results. Among these enriched pathways, the calcium signaling pathway and vascular smooth muscle contraction pathway were of note as they are often reported to be related to regulating the endothelial barrier. Both signaling pathways were shown to be associated with the MLCK-MLC pathway, and the trend shown by proteomics was consistent with relevant literature reports (43–45). In endothelial cells, the calcium signaling pathway is crucial for modulating barrier function and promoting inflammation. It could cause cell-cell junction disassembly and cytoskeletal rearrangement to facilitate endothelial cell retraction and increase permeability (22,23). Moreover, the associated proteins were activated by HS and inhibited by DNAJA1-KO, which is consistent with the aforementioned hypothesis. At the same time, it was found that the MLCK-MLC pathway was highly related to the regulation of vascular endothelial barrier under heat stress through sufficient literature research (46–48). The pivotal protein, MLCK, a key effector of barrier dysfunction, was verified by western blotting. As aforementioned, the elevated expression of MLCK activated MLC and increased the expression of p-MLC in CON + HS cells, which may cause remodeling of the actin cytoskeleton, leading to endothelial hyperpermeability. The present study demonstrated that DNAJA1-KO could inhibit HS-induced MLCK expression and block the subsequent MLC phosphorylation. Taken together, these data confirmed that HS-induced endothelial barrier disruption was mitigated in DNAJA1-KO cells, as indicated by the results of functional tests, and TJ and actin cytoskeleton observations. Moreover, DNAJA1-KO could improve the thermal tolerance of HUVECs, as indicated by cell viability and apoptosis assays. Regarding the mechanism underlying the effects of DNAJA1-KO on the HS-damaged barrier, involvement of the MLCK-MLC signaling pathway was demonstrated from the perspective of endothelial barrier regulation, but could not explain the complete regulatory pathways of DNAJA1-KO-mediated thermal tolerance effects. Although, it was successfully confirmed that DNAJA1KO has a significant protective effect against heat stress injury and HS in vivo and in vitro, DNAJA1 overexpression cell lines must be constructed in future work to fully clarify the mechanisms of the gene action in the future.
In conclusion, DNAJA1-KO could effectively protect the endothelial barrier against HS via improving thermal tolerance and suppressing the MLCK-MLC signaling pathway. These findings emphasize the distinct effects of HSPs on HS and provide a novel insight into the classic heat shock response, which could assist with the identification of therapeutic targets against HS.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This research was funded by the National Key Basic Research (grant no. 2022-JCJQ-ZD-097-11) and the Shanghai Military Civilian Integration Industry Development Project (grant no. 2019-jmrh1-kj52).
Availability of data and materials
The raw data of proteomic analysis generated in the present study may be found in the ProteomeXchange under accession number PXD043512 or at the following URL (https://www.ebi.ac.uk/pride/archive/projects/PXD043512). The remaining data generated in the present study may be requested from the corresponding author.
Authors' contributions
JKC, SGX and JBZ designed the study and, analyzed and interpreted data. JKC and SGX confirm the authenticity of all the raw data. LL, YWW, XC, JLC, MW, JQZ, JFL, LJR, XYD, LY, QS and XCF conducted the experiments and analyzed the data. LL, YWW and XC wrote the article. JKC, SGX, QS and LL revised the article. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by the Committee on Ethic of Medical Research (The Institutional Animal Ethics Committee), Naval Medical University (Shanghai, China; approval no. NMUMREC-2021-002).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
Bouchama A, Abuyassin B, Lehe C, Laitano O, Jay O, O'Connor FG and Leon LR: Classic and exertional heatstroke. Nat Rev Dis Primers. 3:82022. View Article : Google Scholar : PubMed/NCBI | |
|
Epstein Y and Yanovich R: Heatstroke. N Engl J Med. 380:2449–2459. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Raymond C, Matthews T and Horton RM: The emergence of heat and humidity too severe for human tolerance. Sci Adv. 6:eaaw18382020. View Article : Google Scholar : PubMed/NCBI | |
|
Fujimoto M and Nishiura H: Baseline scenarios of heat-related ambulance transportations under climate change in Tokyo, Japan. PeerJ. 10:e138382022. View Article : Google Scholar : PubMed/NCBI | |
|
Aird WC: Endothelium as an organ system. Crit Care Med. 32:S271–S279. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Cheng JL and MacDonald MJ: Effect of heat stress on vascular outcomes in humans. J Appl Physiol (1985). 126:771–781. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Iba T, Connors JM, Levi M and Levy JH: Heatstroke-induced coagulopathy: Biomarkers, mechanistic insights, and patient management. EClinicalMedicine. 44:1012762022. View Article : Google Scholar : PubMed/NCBI | |
|
Xu Q, Liu J, Wang Z, Guo X, Zhou G, Liu Y, Huang Q and Su L: Heat stress-induced disruption of endothelial barrier function Is via PAR1 signaling and suppressed by Xuebijing injection. PLoS One. 10:e01180572015. View Article : Google Scholar : PubMed/NCBI | |
|
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F and Shao F: Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 526:660–665. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Wang S: Genome-wide CRISPR/Cas9 library screening identified DNAJA1 regulating protein homeostasis in heat stress injury, master's dissertation. Naval medical university; 2023 | |
|
Ergulen E, Becsi B, Csomos I, Fésüs L and Kanchan K: Identification of DNAJA1 as a novel interacting partner and a substrate of human transglutaminase 2. Biochem J. 473:3889–3901. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Qiu XB, Shao YM, Miao S and Wang L: The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci. 63:2560–2570. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Kurop MK, Huyen CM, Kelly JH and Blagg BSJ: The heat shock response and small molecule regulators. Eur J Med Chem. 226:1138462021. View Article : Google Scholar : PubMed/NCBI | |
|
Joshi AD, Dimitropoulou C, Thangjam G, Snead C, Feldman S, Barabutis N, Fulton D, Hou Y, Kumar S, Patel V, et al: Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am J Respir Cell Mol Biol. 50:170–179. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Ren Y, Yu G, Shi C, Liu L, Guo Q, Han C, Zhang D, Zhang L, Liu B, Gao H, et al: Majorbio cloud: A one-stop, comprehensive bioinformatic platform for multiomics analyses. iMeta. 1:e122022. View Article : Google Scholar | |
|
Health NIO, . Guide for the care and use of laboratory animals. National Academies; 1985 | |
|
Li L, Man W, Chen J, Xu Z, Wang S, Xia X, Liu D, Wang S, Xie C, Wu J, et al: Preventive effects of bacillus licheniformis on heat stroke in rats by sustaining intestinal barrier function and modulating gut microbiota. Front Microbiol. 12:5482021. | |
|
Gu ZT, Li L, WU F, Zhao P, Yang H, Liu YS, Geng Y, Zhao M and Su L: Heat stress induced apoptosis is triggered by transcription-independent p53, Ca2+ dyshomeostasis and the subsequent Bax mitochondrial translocation. Sci Rep. 5:114972015. View Article : Google Scholar : PubMed/NCBI | |
|
Gu ZT, Wang H, Li L, Liu YS, Deng XB, Huo SF, Yuan FF, Liu ZF, Tong HS and Su L: Heat stress induces apoptosis through transcription-independent p53-mediated mitochondrial pathways in human umbilical vein endothelial cell. Sci Rep. 4:44692015. View Article : Google Scholar | |
|
Zhang S, Liu Y, Wang Z, Liu J, Gu Z, Xu Q and Su L: PAR1-mediated c-Jun activation promotes heat stress-induced early stage apoptosis of human umbilical vein endothelial cells. Mol Med Rep. 15:2595–2603. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Wang Y, Yang C, Elsheikh N, Elsheikh NAH, Li C, Yang F, Wang G and Li L: HO-1 reduces heat stress-induced apoptosis in bovine granulosa cells by suppressing oxidative stress. Aging (Albany NY). 11:5535–5547. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Dalal PJ, Muller WA and Sullivan DP: Endothelial cell calcium signaling during barrier function and inflammation. Am J Pathol. 190:535–542. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Claesson-Welsh L, Dejana E and McDonald DM: Permeability of the endothelial barrier: Identifying and reconciling controversies. Trends Mol Med. 27:314–331. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Clarke JP and Mearow KM: Cell stress promotes the association of phosphorylated HspB1 with F-actin. PLoS One. 8:e689782013. View Article : Google Scholar : PubMed/NCBI | |
|
Yin B, Tang S, Xu J, Sun J, Zhang X, Li Y and Bao E: CRYAB protects cardiomyocytes against heat stress by preventing caspase-mediated apoptosis and reducing F-actin aggregation. Cell Stress Chaperones. 24:59–68. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Dzwinel W, Boryczko K and Yuen DA: A discrete-particle model of blood dynamics in capillary vessels. J Colloid Interface Sci. 258:163–173. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Joffre J and Hellman J: Oxidative Stress and endothelial dysfunction in sepsis and acute inflammation. Antioxid Redox Signal. 35:1291–1307. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Iba T, Helms J, Levi M and Levy JH: Thromboinflammation in acute injury: Infections, heatstroke, and trauma. J Thromb Haemost. 22:7–22. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Liu Z, Zhong T, Zheng D, Cepinskas I, Peng T and Su L: Heat stress pretreatment decreases lipopolysaccharide-induced apoptosis via the p38 signaling pathway in human umbilical vein endothelial cells. Mol Med Rep. 14:1007–1013. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Huang W, Mao L, Xie W, Cai S, Huang Q, Liu Y and Chen Z: Impact of UCP2 depletion on heat stroke-induced mitochondrial function in human umbilical vein endothelial cells. Int J Hyperthermia. 39:287–296. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Wu F, Dong XJ, Zhang HQ, Li L, Xu QL, Liu ZF, Gu ZT and Su L: Role of MnSOD in propofol protection of human umbilical vein endothelial cells injured by heat stress. J Anesth. 30:410–419. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Liu Y, Zhou G, Wang Z, Guo X, Xu Q, Huang Q and Su L: NF-κB signaling is essential for resistance to heat stress-induced early stage apoptosis in human umbilical vein endothelial cells. Sci Rep. 5:135472015. View Article : Google Scholar : PubMed/NCBI | |
|
Antonov A, Snead C, Gorshkov B, Antonova GN, Verin AD and Catravas JD: Heat shock protein 90 inhibitors protect and restore pulmonary endothelial barrier function. Am J Respir Cell Mol Biol. 39:551–559. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Yuan X, Chen Y, Chen G, Liu G, Hang M, Wang P, Luo Y, Guo D and Xu L: The heat shock protein 70 plays a protective role in sepsis by maintenance of the endothelial permeability. Biomed Res Int. 2020:21940902020. View Article : Google Scholar : PubMed/NCBI | |
|
Bi J, Zhang J, Ren Y, Du Z, Zhang Y, Liu C, Wang Y, Zhang L, Shi Z, Wu Z, et al: Exercise hormone irisin mitigates endothelial barrier dysfunction and microvascular leakage-related diseases. JCI Insight. 5:e1362772020. View Article : Google Scholar : PubMed/NCBI | |
|
Wang Z, Lu YL, Chen M, Xu HF and Zheng LR: Piceatannol alleviates glucolipotoxicity induced vascular barrier injury through inhibition of the ROS/NF-kappa B signaling pathway. Am J Transl Res. 14:120–134. 2022.PubMed/NCBI | |
|
Du L, Dong F, Guo L, Hou Y, Yi F, Liu J and Xu D: Interleukin-1beta increases permeability and upregulates the expression of vascular endothelial-cadherin in human renal glomerular endothelial cells. Mol Med Rep. 11:3708–3714. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Hohmann T and Dehghani F: The cytoskeleton-A complex interacting meshwork. Cells. 8:3622019. View Article : Google Scholar : PubMed/NCBI | |
|
Liu R, Niu X, Yuan J, Chen H, Gao X and Qi R: DnaJA4 is involved in responses to hyperthermia by regulating the expression of F-actin in HaCaT cells. Chin Med J (Engl). 134:456–462. 2021. View Article : Google Scholar | |
|
Wettstein G, Bellaye PS, Micheau O and Bonniaud P: Small heat shock proteins and the cytoskeleton: An essential interplay for cell integrity? Int J Biochem Cell Biol. 44:1680–1686. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Almeida-Souza L, Asselbergh B, D'Ydewalle C, Moonens K, Goethals S, de Winter V, Azmi A, Irobi J, Timmermans JP, Gevaert K, et al: Small heat-shock protein HSPB1 mutants stabilize microtubules in charcot-marie-tooth neuropathy. J Neurosci. 31:15320–15328. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Kayser J, Haslbeck M, Dempfle L, Krause M, Grashoff C, Buchner J, Herrmann H and Bausch AR: The small heat shock protein Hsp27 Affects assembly dynamics and structure of keratin intermediate filament networks. Biophys J. 105:1778–1785. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Du L, Zhu L, Lu X, Yu Y, Liu P and Pan J: Inhibition of the MLCK/MLC2 pathway protects against intestinal heat stroke-induced injury in rats. J Therm Biol. 116:1036552023. View Article : Google Scholar : PubMed/NCBI | |
|
Yang PC, He SH and Zheng PY: Investigation into the signal transduction pathway via which heat stress impairs intestinal epithelial barrier function. J Gastroenterol Hepatol. 22:1823–1831. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Li H, Chen H, Zhang S, Wang S, Zhang L, Li J, Gao S and Qi Z: Taurine alleviates heat stress-induced mammary inflammation and impairment of mammary epithelial integrity via the ERK1/2-MLCK signaling pathway. J Therm Biol. 116:1035872023. View Article : Google Scholar : PubMed/NCBI | |
|
Cheng M, Yang Z, Qiao L, Yang Y, Deng Y, Zhang C and Mi T: AGEs induce endothelial cells senescence and endothelial barrier dysfunction via miR-1-3p/MLCK signaling pathways. Gene. 851:1470302023. View Article : Google Scholar : PubMed/NCBI | |
|
Zhong J, Yu R, Zhou Q, Liu P, Liu Z and Bian Y: Naringenin prevents TNF-alpha-induced gut-vascular barrier disruption associated with inhibiting the NF-kappaB-mediated MLCK/p-MLC and NLRP3 pathways. Food Funct. 12:2715–2725. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Huang Y, Luo X, Li X, Song X, Wei L, Li Z, You Q, Guo Q and Lu N: Wogonin inhibits LPS-induced vascular permeability via suppressing MLCK/MLC pathway. Vascul Pharmacol. 72:43–52. 2015. View Article : Google Scholar : PubMed/NCBI |