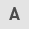Compound 225# inhibits the proliferation of human colorectal cancer cells by promoting cell cycle arrest and apoptosis induction
- Authors:
- Published online on: April 2, 2024 https://doi.org/10.3892/or.2024.8729
- Article Number: 70
-
Copyright: © Zhang et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Colorectal cancer (CRC) is one of the most dangerous human malignant tumors, and it is the third most prevalent cancer diagnosed in both men and women in the United States. According to the American Cancer Society, there will be 106,970 new cases of colon cancer and 46,050 new cases of rectal cancer in the United States in 2023 (1). CRC is common, particularly in economically developed nations, and is associated with lifestyle changes, such as current eating habits, an increase in smoking prevalence, a lack of exercise, and an increase in the number of individuals that are overweight and obese (2). CRC has a high chance of recurrence, a low postoperative 5-year survival rate and a high rate of metastasis (3). Chemotherapeutic drugs are the most commonly used therapy for patients with CRC in numerous circumstances. These therapies, however, cause a number of dose-limiting adverse effects, including delayed diarrhea, gastrointestinal mucositis, neutropenia and myelosuppression (4). As a result, the development of new effective antineoplastic medicines with fewer side effects remains critical.
It has been demonstrated that cell cycle arrest depends on the activation of DNA damage signaling pathways, which also influence the proliferation of cancer cells (5). Previous studies have reported that selectively targeting specific biomarkers involved in the regulation of cell division or apoptosis can have a significant impact on tumor growth or the supportive environment of cancer cells, while having minimal influence on normal cells (6–8). The identification of compounds that can inhibit the cell cycle and induce apoptosis shows promise in the development of potential drugs for CRC therapy. Numerous studies have indicated that endoplasmic reticulum (ER) stress plays a vital role as a molecular mechanism in cancer therapy. Upon drug stimulation, unfolded proteins can accumulate in the cytoplasm, which can then lead to the initiation of ER stress (9,10). Persistent ER stress activates the PERK downstream signal eIF2α, resulting in the suppression of protein translation; this ultimately leads to cellular damage and apoptosis (11).
The present study synthesized a novel small molecule compound, 1-[(4-ethoxyphenyl) amino]-7a,11a-dihydro-3H-naphtho [1,2,3-de] quinoline-2,7-dione (compound 225#), and explored whether compound 225# could inhibit the proliferation of CRC both in vitro and in vivo. The present study further discussed the mechanism, in order to determine whether it could be considered a potential candidate for CRC therapy or adjuvant therapy.
Materials and methods
Reagents and antibodies
Primary antibodies against P21 (cat. no. 2947T), cyclin B1 (cat. no. 4138T), phosphorylated (p)-checkpoint kinase 2 (CHK2) (cat. no. 2197T), CHK2 (cat. no. 2662T), p-ATR (cat. no. 2853T), ATR (cat. no. 2790S), p-BRCA1 (cat. no. 9009T), BRCA1 (cat. no. 9010T), γ-H2A.X (cat. no. 9718T), H2A.X (cat. no. 7631T), p-P53 (cat. no. 9286T), P53 (cat. no. 9282T), P27 (cat. no. 2552T), S-phase kinase-associated protein 2 (SKP2; cat. no. 4358T), PARP (cat. no. 9542T), PUMA (cat. no. 4976T), calnexin (cat. no. 2679T), protein disulfide-isomerase (PDI; cat. no. 3501T), p-eIF2α (cat. no. 3398T), eIF2α (cat. no. 5324T), PERK (cat. no. 5683T), ubiquitin (Ub; cat. no. 20326T) and LC3B (cat. no. 3868T) were purchased from Cell Signaling Technology, Inc.; primary antibodies against α-tubulin (cat. no. AF0001) and CDK1 (cat. no. AF0111) were purchased from Beyotime Institute of Biotechnology; and primary antibodies against cyclin A1 (cat. no. 13295-1-AP) were purchased from Proteintech Group, Inc. All of the primary antibodies were diluted to 1:1,000 with western primary antibody diluent (cat. no. AZ100; Beyotime Institute of Biotechnology). The anti-rabbit IgG (DyLight™ 800 4X PEG Conjugate; cat. No. 5151P) and anti-mouse IgG (DyLight™ 680 Conjugate; cat. no. 5470P) secondary antibodies were obtained from Cell Signaling Technology, Inc. (1:15,000). MTT (cat. no. ST316), BeyoClick™ EdU-594 Cell proliferation Detection Kit (cat. no. C0078S), 0.5% crystal violet (cat. no. C0121), propidium iodide (PI; cat. no. ST512), RNase A (cat. no. ST578), Annexin V-FITC (cat. no. C1062S) and BCA kit (cat. no. P0010) were purchased from Beyotime Institute of Biotechnology. Dimethyl sulfoxide (DMSO; cat. no. D2650) and polyvinylidene difluoride (PVDF) membranes were purchased from MilliporeSigma.
Synthesis of compound 225#
The synthesis of compound 225# is shown in Fig. 1. Chloroacetyl chloride (1.5 eq) was added to a stirring suspension of 1-aminoanthraquinone compound 1 (1 eq) in benzene. The mixture was stirred at 70°C for 12 h. The precipitate was isolated by filtration and washed sequentially with saturated NaHCO3, deionized water and ethanol. The yellow compound 2 was dried under vacuum for 24 h and obtained an 89% yield. Subsequently, a mixture of compound 2 and triethylamine (3 eq) in ethanol was refluxed for 12 h. After the mixture was cooled to room temperature, it was placed at 4°C. The solid precipitate was filtered off, washed with a small amount of cold ethanol and air dried to provide compound 3 (15% yield). To obtain compound 225#, a mixture of compound 3 (1 eq), 4-ethoxyaniline (1 eq) and anhydrous sodium acetate (1 eq) was heated at 190°C for 12 h. After cooling, water was added to the reaction mixture, and the solid was filtered out of the nitrobenzene solution, and after drying and recrystallization, the target compound with a yield of 7% was obtained. 1H and 13C NMR were measured on a Bruker 400 spectrometer (Bruker Corporation) (Fig. S1).
Cell culture
The human colon carcinoma cell lines HCT116 and SW620, as well as the normal human colon cell line FHC and 293T cells were obtained from the American Type Culture Collection. HCT116 cells were cultured in McCoy's 5a medium (cat. no. 16600108) supplemented with 10% fetal bovine serum (FBS; cat. no. 10100147, Australia origin) (both from Gibco; Thermo Fisher Scientific, Inc.). SW620 cells were cultured in high-glucose DMEM (cat. no. SH30022.01; Hyclone; Cytiva) supplemented with 10% FBS. FHC cells were cultured in DMEM: F12K medium (cat. no. 11330032; Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (cat. no. 10100147; Australia origin; Gibco; Thermo Fisher Scientific, Inc.), 10 ng/ml cholera toxin (cat. no. HY-P1446; MedChemExpress), 0.005 mg/ml insulin (cat. no. I9278; MilliporeSigma), 0.005 mg/ml transferrin (cat. no. T3309-1; MilliporeSigma) and 100 ng/ml hydrocortisone (cat. no. H0888-500; MilliporeSigma). All cells were cultured in an incubator at 37°C under a humidified atmosphere of 5% CO2.
Cell viability assay
The cell viability and IC50 value of compound 225# were determined using the MTT assay. HCT116 and SW620 cells were counted and seeded into 96-well plates at a density of 1×103 cells/well with 200 µl complete medium. After a 12-h pre-incubation, the cells were exposed to different concentrations of compound 225# (0, 1.5, 3.1, 6.25, 12.5, 25, 50, 100 and 200 nM) for 1–4 and 5 days at 37°C. To assess cell viability, 20 µl MTT solution (5 mg/ml in PBS) was added to each well and incubated for an additional 4 h. The medium was then removed, and 200 µl DMSO was added to dissolve the formazan crystals. The plate was agitated for 10 min, and then the optical density was measured at an absorbance wavelength of 570 nm using a microplate reader (BioTek Instruments, Inc.). The experiments were repeated three times, and proliferation inhibition curves and IC50 values were generated using GraphPad Prism 5 (Dotmatics) (12).
EdU staining assay
Cell proliferation was assessed using the BeyoClick™ EdU-594 Cell Proliferation Detection kit. CRC cell lines (SW620 and HCT116) were counted and seeded into 96-well plates at a density of 1×104 cells/well with 200 µl complete medium. After 12 h of pre-incubation, SW620 and HCT116 cells were treated with 0, 12.5, 25 and 50 nM compound 225# for 24 h at 37°C. Subsequently, EdU was added to each well and incubated at 37°C for 2 h, followed by staining with Hoechst 33342 for 30 min at room temperature. Analysis was performed using the High Content Analysis System-Operetta CLS™ (PerkinElmer, Inc.).
Colony formation assay
A colony formation assay was used to assess in vitro proliferation of CRC cells following treatment with the indicated concentrations of compound 225#. Briefly, CRC cell lines (SW620 and HCT116) were seeded onto 6-well plates at a density of 1×103 cells/well in 2 ml medium. After 12 h of culture, SW620 and HCT116 cells were treated with 0, 6.5, 12.5 and 25 nM compound 225# for 10 days at 37°C. Subsequently, the cells were washed twice with PBS and fixed with 4% paraformaldehyde for 30 min at room temperature. After another round of washing, the cells were stained with 0.5% crystal violet (cat. no. C0121; Beyotime Institute of Biotechnology) for 15 min at room temperature and examined for colony formation (colonies were defined as >50 cells). Excessive crystal violet was washed away, and the colonies were observed and analyzed using an Epson scanner (Epson Corporation).
Cell cycle analysis
The effect of compound 225# on cell cycle distribution was assessed using a flow cytometer (Accuri™ C6; BD Biosciences) after PI staining. SW620 and HCT116 cells were seeded onto a 6-cm Petri dish at a density of 2×106 cells/dish. After overnight incubation, the cells were treated with different concentrations of compound 225# (0, 25, 50 and 100 nM) for 24 h at 37°C. Subsequently, the cells were fixed in 70% ice-cold ethanol at 4°C for 24 h, resuspended in 200 µl PBS containing 1 µl PI (5 mg/ml) and 1 µl RNase A (5 mg/ml), and incubated in the dark for 30 min at 37°C. The PI fluorescence was measured using flow cytometry and the data were obtained by analyzing with FlowJo7.6 software (FlowJo, LLC) (4).
Flow cytometric analysis of apoptosis
SW620 and HCT116 CRC cells were seeded onto a 6-cm Petri dish (2×106 cells/dish). After overnight culture, the cells were incubated with compound 225# (0, 50, 100 and 200 nM) for 48 h at 37°C. After collecting the cells, they were washed with ice-cold PBS, and then resuspended in 200 µl PBS containing 5 µl Annexin V-FITC and 5 µl PI (50 µg/ml). After incubation in the dark for 15 min at room temperature, the samples were analyzed using a flow cytometer (Accuri™ C6; BD Biosciences) and the data were obtained by analyzing with FlowJo7.6 software.
Fluorescence observation of mCherry-GFP-LC3B
To further investigate whether compound 225# could regulate the progression of autophagic flux, HCT116 cells were infected with a double tagged mCherry-GFP-LC3B reporter, which is pH sensitive. This allowed for the assessment of the fusion efficiency of autophagosomes and lysosomes. Using lentivirus infection, HCT116 cells stably expressing mCherry-GFP-LC3B (cat. no. P4837; Wuhan MiaoLing Biotech Science) were created. The mCherry-GFP-LC3B, Pspax2 (cat. no. P0261; Wuhan MiaoLing Biotech Science) and pMD2G (cat. no. JY03028; Nanjing Jiang Yuan Biotechnology, Co. Ltd.) vectors were co-transfected into 293T cells at a 1:1:1 ratio using the Lipo8000™ Transfection Reagent (cat. no. C0533; Beyotime Institute of Biotechnology). Following a 72-h incubation at 37°C, the lentiviral particles were extracted and subsequently introduced into the medium of HCT116 cells at a multiplicity of infection of ~10, which was augmented with 10 µg/ml polybrene. After 24 h at 37°C, the medium was replaced and 10 µg/ml puromycin was used to select the cells that stably expressed the mCherry-GFP-LC3B vector. Cells were grown overnight within a 6-cm Petri dish (2×106 cells/dish) before treatment with compound 225# (0, 25, 50 and 100 nM) for 48 h at 37°C, after which, they were fixed with 4% paraformaldehyde for 30 min at room temperature, washed several times with PBS and the High Content Analysis System-Operetta CLS™ (PerkinElmer, Inc.) was used to analyze the data.
Western blotting
After being treated with compound 225# (0, 50, 100 and 200 nM) for 24 h at 37°C, SW620 and HCT116 human CRC cells were collected and were added to lysis buffer (cat. no. P0013; Beyotime Institute of Biotechnology) containing protease inhibitor; after 30 min on ice, the samples were centrifuged at 16,602 × g at 4°C for 15 min. A BCA kit was used to assess the protein concentration in the supernatant. Subsequently, equal amounts of protein were separated by 8–15% SDS-PAGE, transferred to PVDF membranes and incubated at room temperature for 1 h with 5% skim milk, followed by overnight incubation at 4°C with primary antibodies. Membranes were rinsed with TBS-0.5% Tween before being incubated with secondary antibodies for 1 h at room temperature. Finally, membranes were visualized on a Tanon 5200 Imaging System (Tanon Science and Technology Co., Ltd.). The gray scale of the strip was analyzed by ImageJ 1 (National Institutes of Health), and the expression levels of the target protein were calculated using α-tubulin as a reference.
Xenograft mouse model
A total of 30 nude Balb/c female mice (age, 4–6 weeks; weight, ~20 g) were obtained from Hunan SJA Laboratory Animal Co., Ltd. The mice were cultured in a specific pathogen-free animal room in ventilated cages with a temperature of 28°C and humidity of 50%, under 10 h of light and 14 h of dark, and were provided ad libitum access to sufficient food and water. The experimental procedures were conducted with the animals under 2.5% isoflurane gas anesthesia. Subcutaneous injection of HCT116 cells (5×106 cells) was performed on the nude mice to establish the xenograft model. When tumors reached an average volume of ~100 mm3, 30 mice were divided randomly into three groups (n=10/group), as follows: Control group (0 mg/kg), and 10 and 30 mg/kg compound 225# groups. The compound 225# groups received intragastric administration with 10 or 30 mg/kg compound 225# dissolved in 100 µl solvent (5% DMSO, 30% PEG300, 10% Tween-80 and 55% saline). The drug administration course was set for 6 days (once a day for 3 consecutive days, followed by three days of withdrawal), and five courses were performed. Tumor length (L) and width (W) were measured using a vernier caliper every 3 days, and the tumor volume was calculated using the standard formula: (L × W2)/2. After 32 days, all mice were euthanized by cervical dislocation, and the tumors were excised and weighed for further analysis.
Statistical analysis
All data were analyzed using GraphPad Prism 5.0 and SPSS 18.0 (SPSS, Inc.) and are presented as the mean ± SD of at least three independent experiments. Statistical analysis was performed using one-way ANOVA for comparison of more than two groups followed by Tukey's post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Compound 225# inhibits the proliferation of CRC cells in vitro
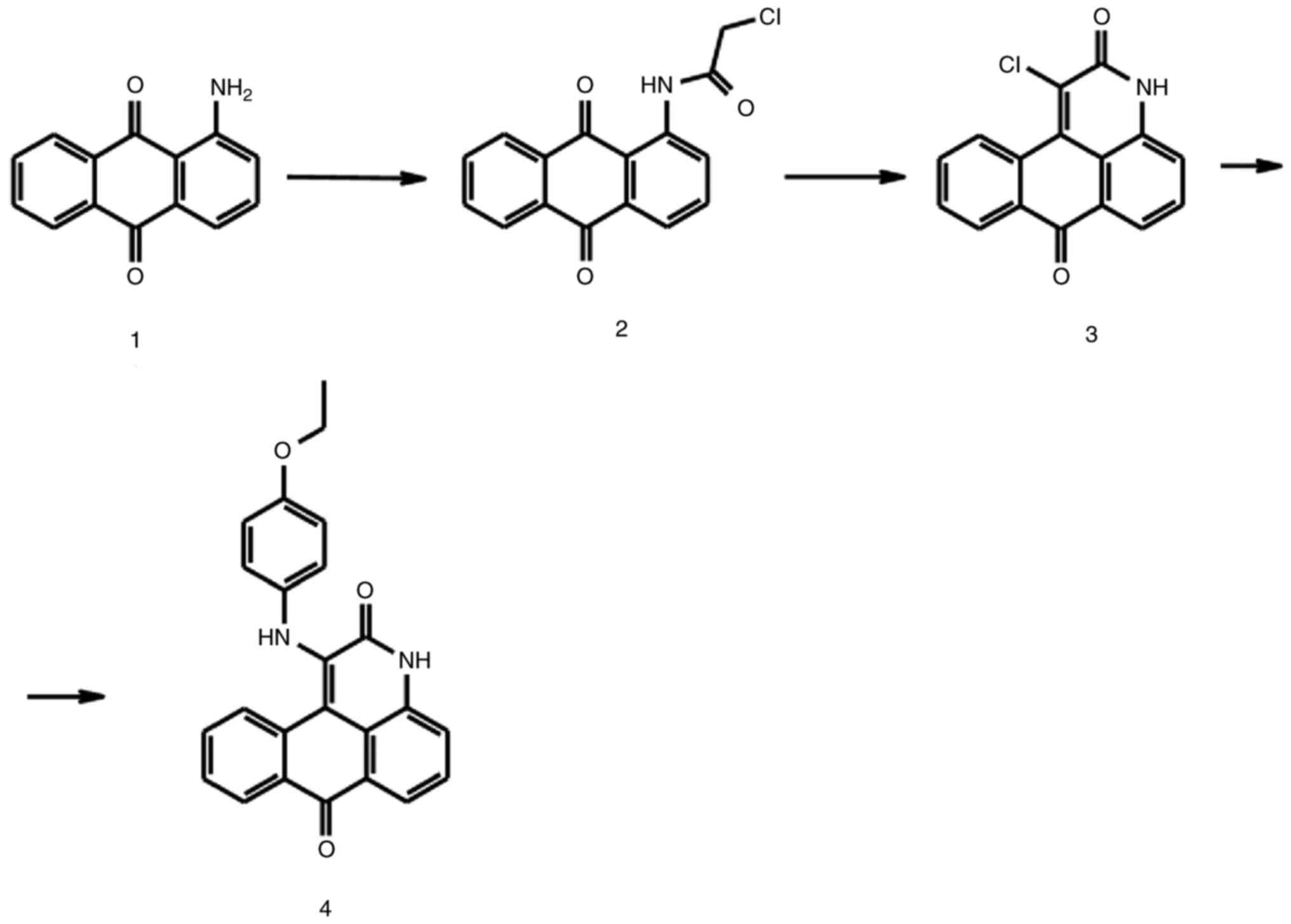
To investigate the anti-proliferative activity of compound 225# on human CRC cells, HCT116 and SW620 cells were treated with compound 225# for 1–4 and 5 days, and cell viability was measured by MTT assay. IC50 values of compound 225# for HCT116 and SW620 cells are presented in Table I. As shown in Fig. 2A, the viability of human CRC cells was decreased in a dose- and time-dependent manner. Moreover, compound 225# had little effect on human rectal mucosal cells (FHC) at the same concentrations (Fig. 2A), indicating its low toxicity to normal cells. Similarly, the colony formation assay demonstrated that compound 225# was able to effectively reduce the size and number of colonies in a dose-dependent manner (Fig. 2B). Compared with that in the control group, the number of EdU-positive cells was significantly reduced in a dose-dependent manner after being exposed to compound 225# (Fig. 2C), thus indicating that compound 225# has the ability to inhibit the proliferation of HCT116 and SW620 cells. These results suggested that compound 225# acts as an actual inhibitor to suppress the proliferation of human CRC cells.
Compound 225# induces cell cycle arrest at G2/M phase
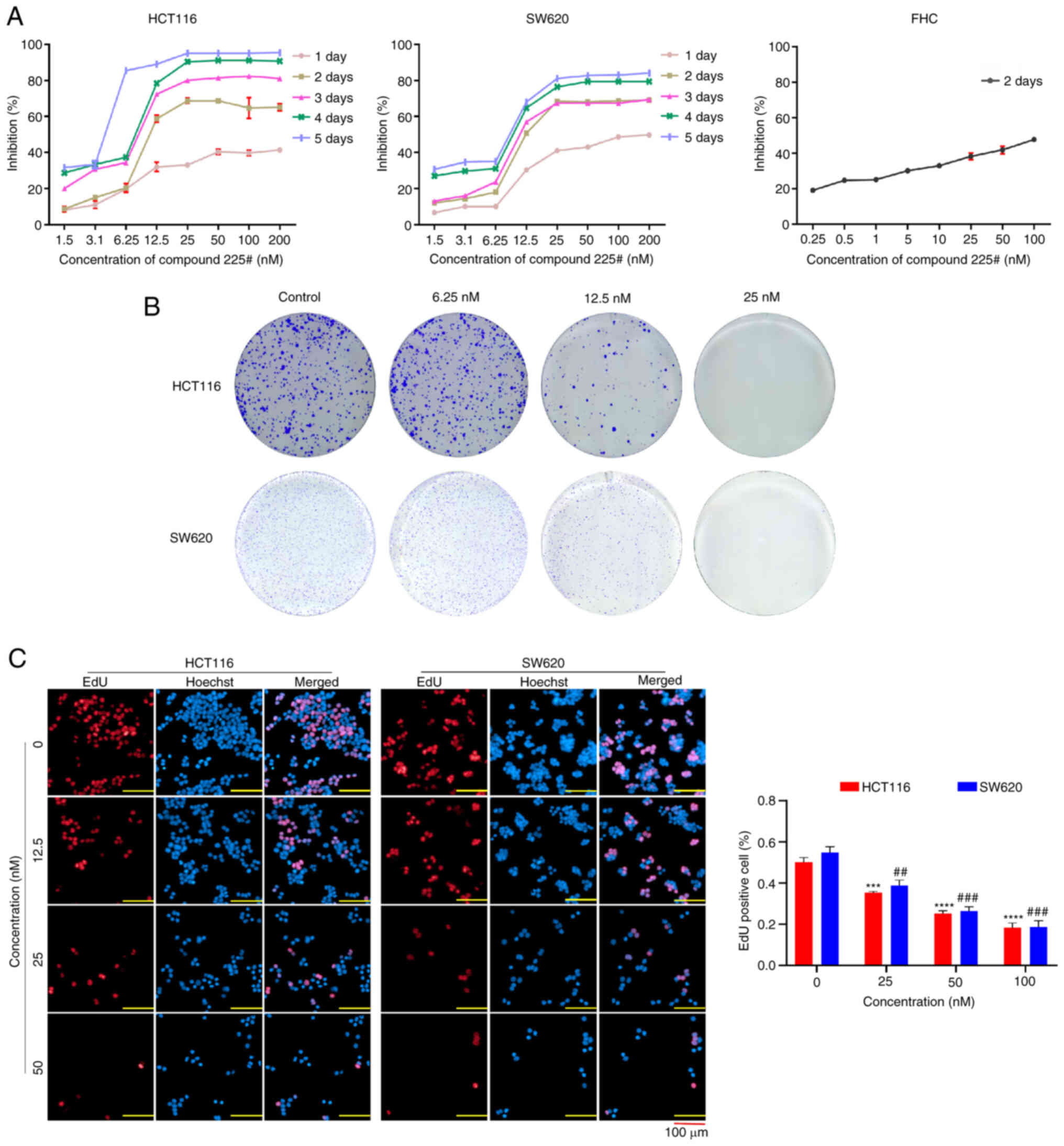
To further clarify the mechanism underlying the anti-CRC effect of compound 225#, the present study investigated its impact on cell cycle progression. Flow cytometry showed that compound 225# induced a significant dose-dependent increase in the number of cells in G2/M phase after 24 h of incubation (Fig. 3A). Notably, 100 nM compound 225# increased the number of SW620 cells in G2/M phase by 68.8% and reduced the number of cells in S phase by 36.4% compared with that in the control group. Similarly, treatment with 100 nM compound 225# induced a significant increase (62.3%) in the number of HCT116 cells in G2/M phase and an obvious reduction (21.3%) in the number of cells in S phase compared with that in the control (Fig. 3A). To further evaluate its effects on the progression of mitosis, the expression levels of cell cycle-related proteins were analyzed via western blotting. The expression levels of cyclin A1, cyclin B1, CDK1 and SKP2 were reduced in response to compound 225# compared with those in the control group, whereas the expression levels of P21 and P27 were increased in a dose-dependent manner (Fig. 3B). Based on these data, compound 225# may suppress human CRC cell proliferation by regulating key molecules in G2/M phase.
It has been reported that cell cycle arrest depends on activation of the DNA damage pathway (4). Hence, DNA damage-related proteins were detected using western blotting to evaluate whether DNA damage was present in CRC cells. As expected, the expression levels of p-CHK2, p-ATR, p-BRCA1 and γ-H2A.X were upregulated, whereas p-P53 was slightly downregulated in both HCT116 and SW620 cells after treatment with compound 225# (Fig. 3C). Therefore, these results indicated that compound 225# may be capable of causing DNA damage in CRC cells, thus resulting in cell cycle arrest in G2/M phase.
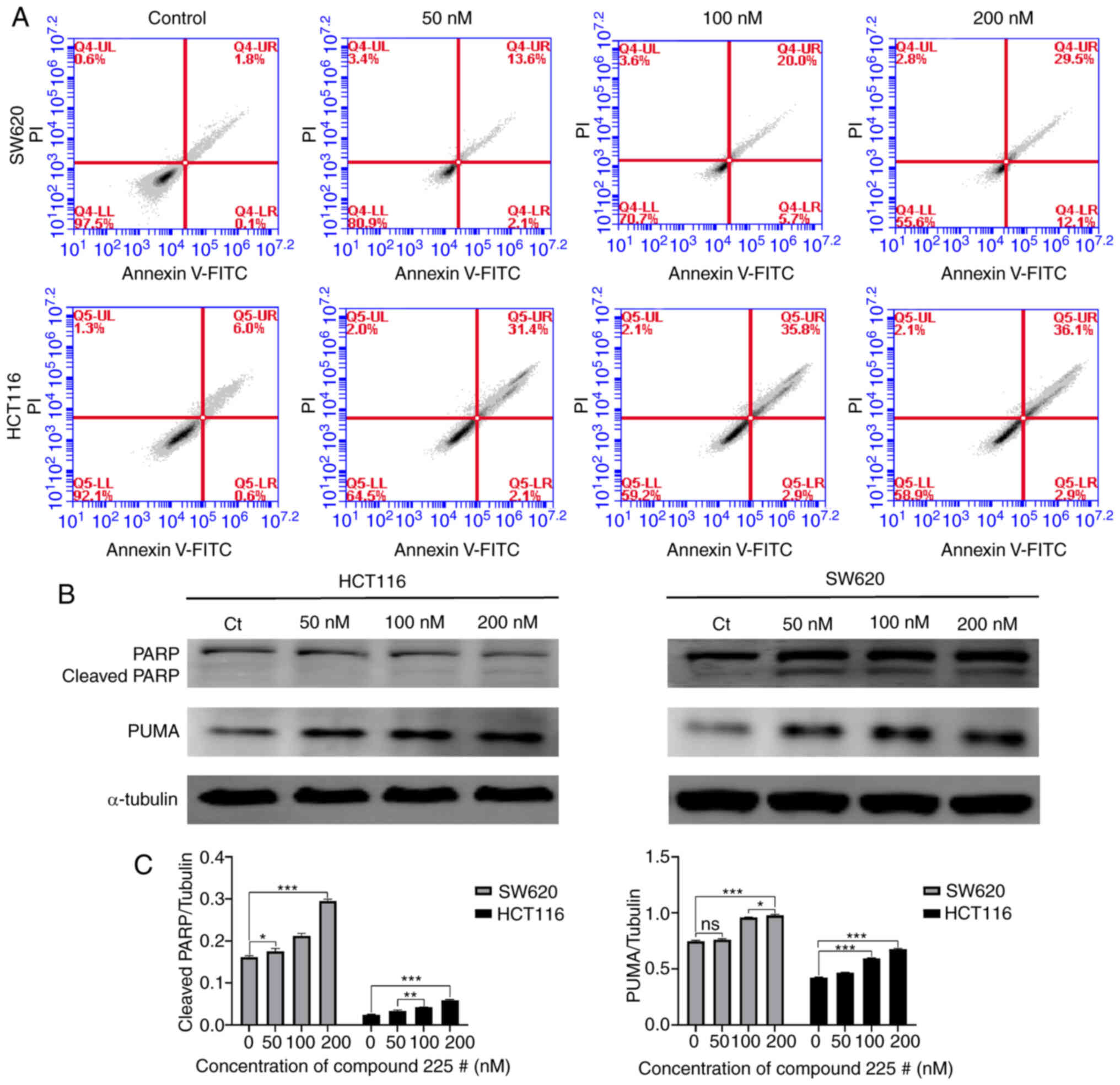
Compound 225# induces apoptosis in human CRC cells
The present study investigated whether apoptosis was triggered in response to compound 225#. The results of the flow cytometric analysis indicated that compound 225# induced apoptosis in both SW620 and HCT116 cells, and increased the proportion of cells in late-phase apoptosis (from 1.8 to 29.5% for SW620 cells; from 6 to 36.1% for HCT116 cells) in a concentration-dependent manner (Fig. 4A). Subsequently, the expression levels of apoptosis-related proteins were assessed by western blotting. As expected, the expression levels of cleaved PARP were increased in response to compound 225# treatment (Fig. 4B). Notably, the expression levels of PUMA were also enhanced in both SW620 and HCT116 cells following treatment with compound 225# (Fig. 4B). In summary, these data suggested that compound 225# may have the ability to regulate the activity of apoptosis-related proteins, thereby inducing apoptosis in human CRC cells.
Compound 225# induces ER stress response and ubiquitinated aggregates accumulate in CRC cells
In general, when cells are stimulated by external factors, such as cytoviral infection and cellular nutrient deficiencies, a significant accumulation of unfolded or misfolded proteins occurs in the ER lumen, this accumulation eventually leads to ER stress and subsequent cell apoptosis (13). It was hypothesized that ER stress may be activated after exposure to compound 225#, due to the detected induction of apoptosis. Therefore, the effect of accumulated intracellular misfolded proteins on ER stress was investigated using western blotting. As shown in Fig. 5A, the expression levels of ER stress-related proteins, including p-eIF2α, PDI, PERK and calnexin were increased in a dose-dependent manner. These findings suggested that treatment with compound 225# may result in the induction of unfolded or misfolded protein accumulation.
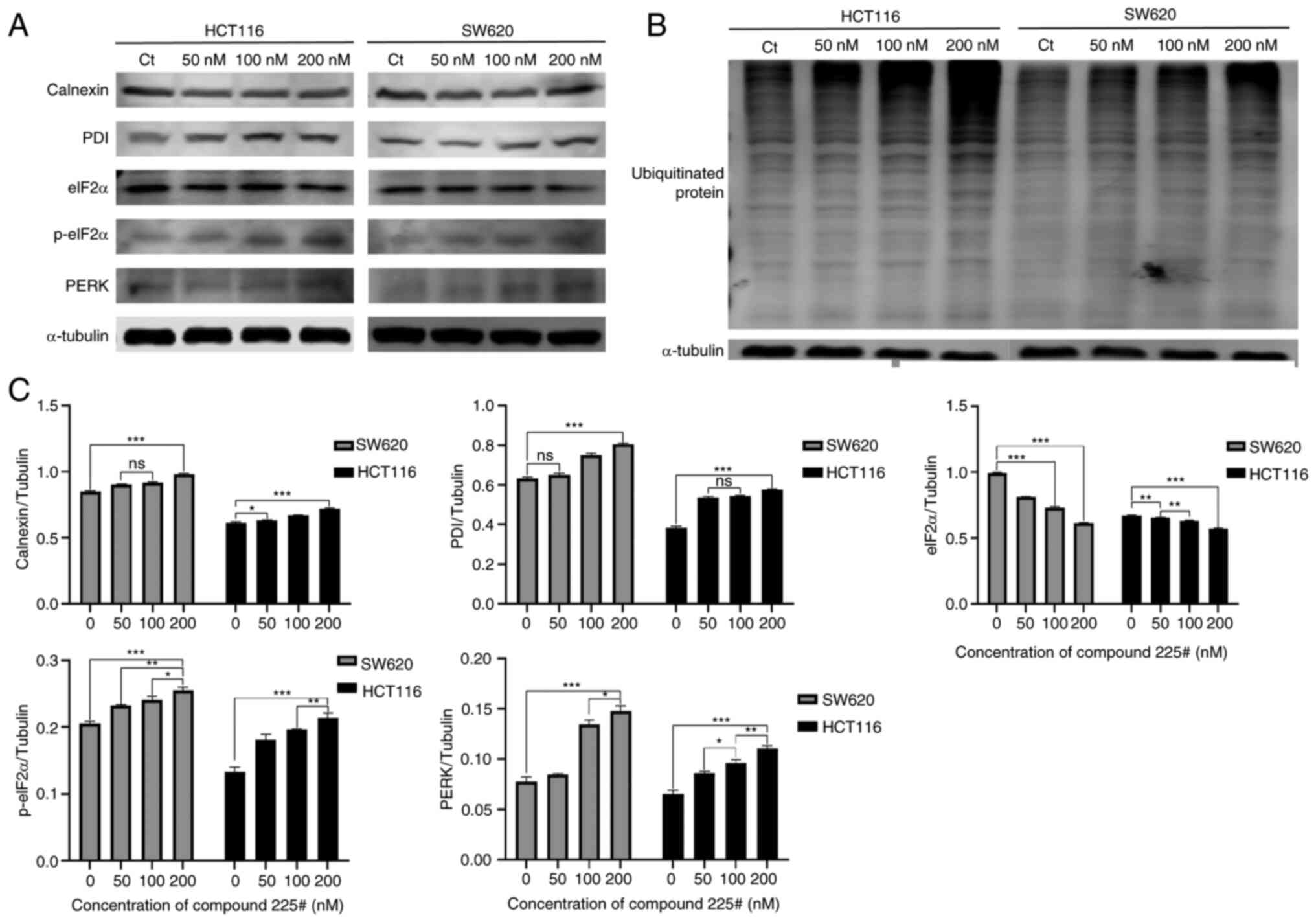
It has been reported that unfolded or misfolded proteins will be tagged by Ub before stimulating ER stress (14); therefore, the present study investigated whether ubiquitinated proteins were aggregated following compound 225# treatment. As shown in Fig. 5B, the accumulation of ubiquitinated aggregates showed a dose-dependent increase following treatment with compound 225#. In summary, these observations demonstrated that compound 225# induced the accumulation of ubiquitinated aggregates, which could further initiate ER stress.
Compound 225# induces autophagy in human CRC cells
Multiple studies have provided evidence that ER stress can induce the activation of autophagy, leading to the engulfment of stressed ER through the formation of autophagosomes (15). To determine whether compound 225# could regulate autophagy, western blotting was performed to detect the change in LC3-II after treatment with the inhibitor. Notably, compound 225# induced an accumulation of LC3B-II in a dose-dependent manner in both SW620 and HCT116 cells (Fig. 6A), indicating that autophagy was activated.
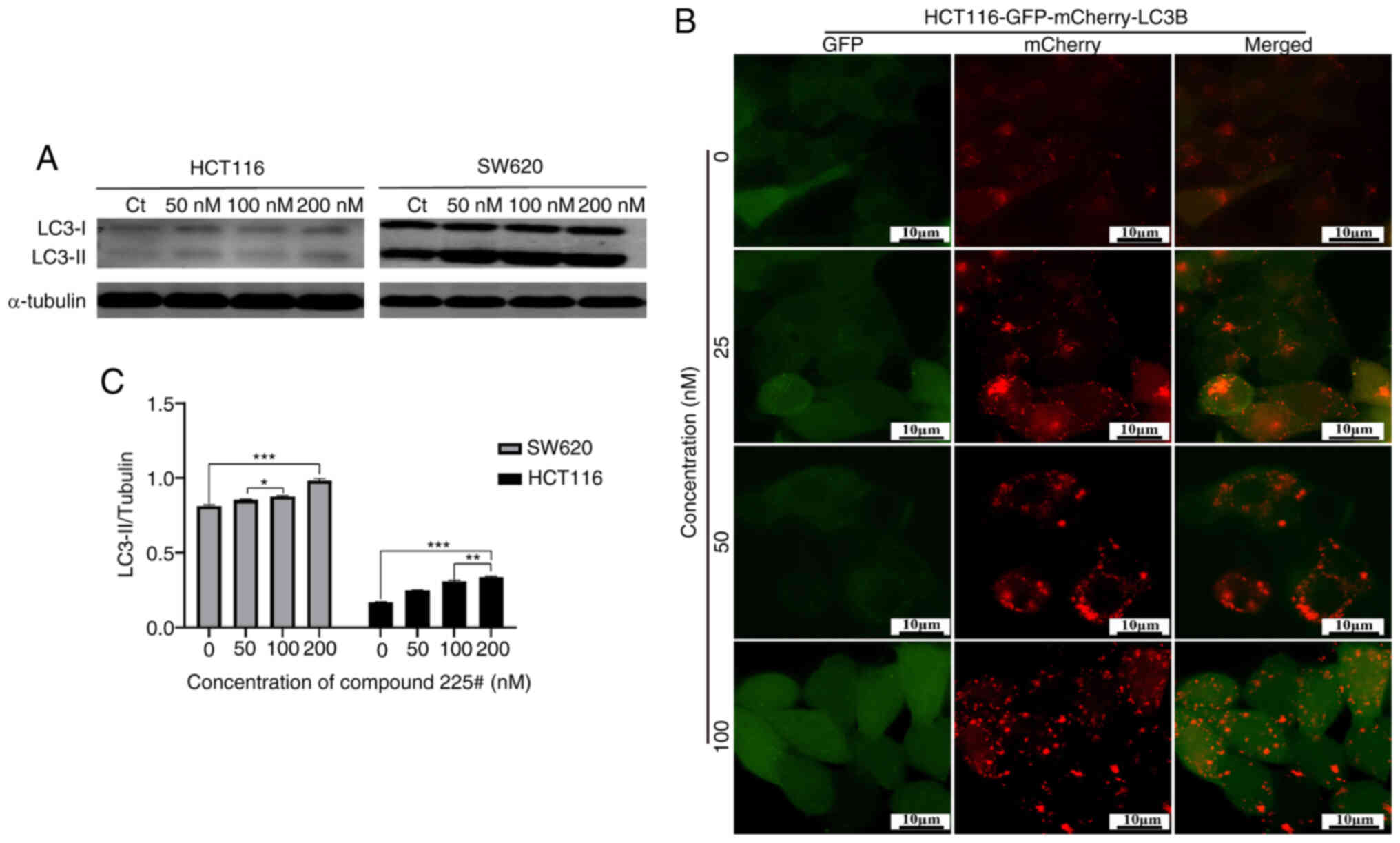
The regulation of autophagic flux by compound 225# was shown in Fig. 6B; compared with in the control group, red fluorescent dots were more pronounced and almost no yellow fluorescent dots were produced in compound 225#-treated HCT116 cells, suggesting that the acidic environment of lysosomes led to the quenching of GFP fluorescence after treatment with compound 225#, and fusion of autophagosomes with lysosomes took place, indicating the promotion of autophagic flux.
Compound 225# inhibits CRC tumor growth in vivo
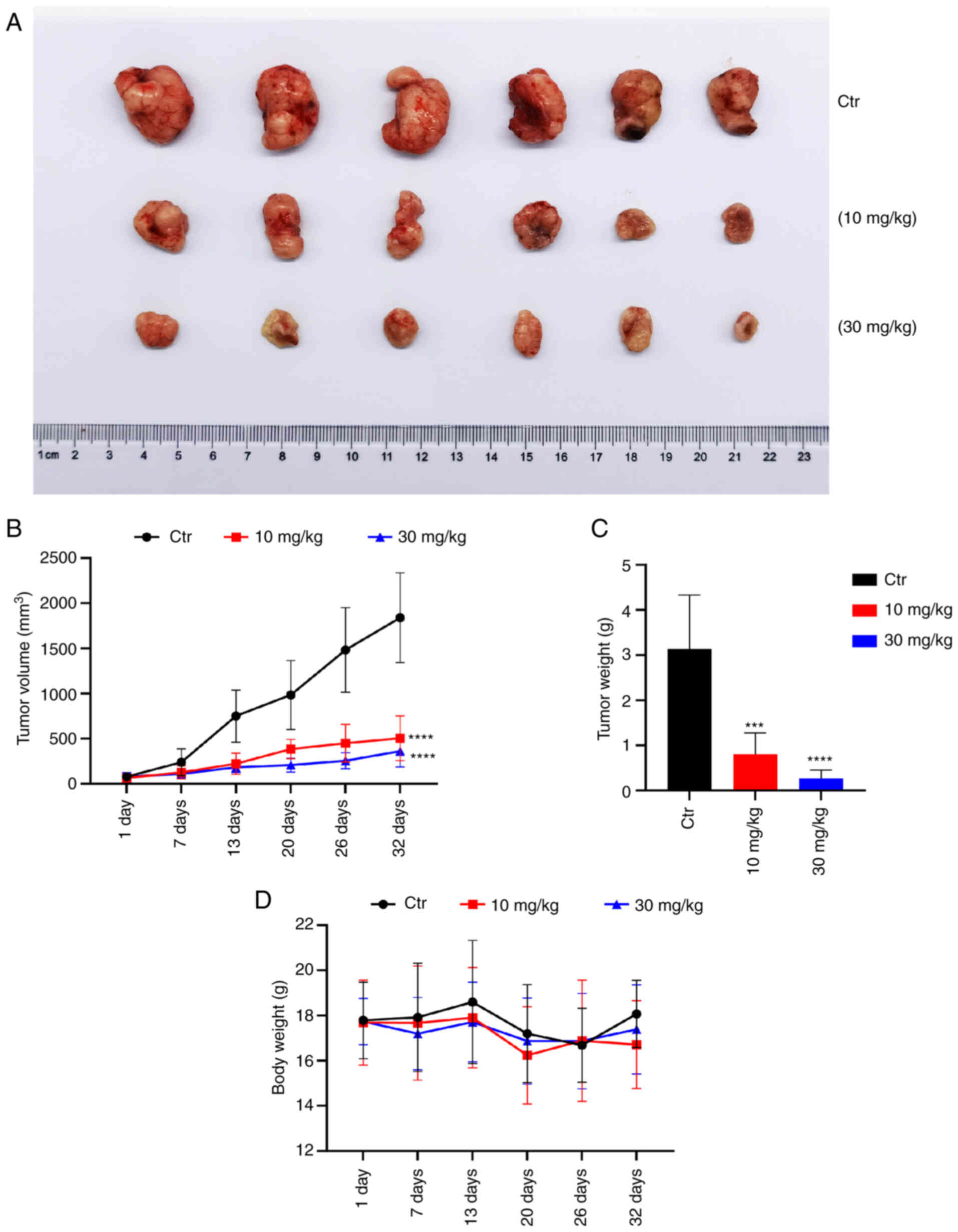
To investigate the effect of compound 225# on CRC tumor growth in vivo, an HCT116 ×enograft mouse model was established. As shown in Fig. 7A, the administration of compound 225# for 32 days markedly suppressed tumor growth in mice bearing HCT116 tumors. After 6 days of treatment, the average tumor volume in the groups treated with 10 and 30 mg/kg compound 225# was significantly smaller than that in the control group (Fig. 7B). By day 32, the average tumor volume in 10 and 30 mg/kg compound 225#-treated groups was only 27.4 and 14.3% of that in the control group, respectively. Moreover, the weights of tumors in the 10 and 30 mg/kg compound 225#-treated groups were reduced by 72.7 and 90.6%, respectively, compared with that in the control group (Fig. 7C). Additionally, at day 32, mice treated with compound 225# showed a decrease in body weight compared with that in the control group, but it was not significant (Fig. 7D). Overall, these results indicated that compound 225# may effectively inhibit HCT116 CRC tumor growth in vivo in an in vivo setting.
Discussion
CRC is a prevalent malignant tumor affecting the digestive system, with its global incidence ranking second only to lung cancer among malignant tumors (2). Chemotherapy is a crucial component in the treatment and prognosis of patients with CRC. However, the current chemotherapeutics have limitations in terms of their effectiveness, due to side effects and the emergence of drug resistance. Consequently, it is imperative to explore and identify new drugs that possess greater potency and lower toxicity levels. In the present study, compound 225# was synthesized and exhibited some extent of anti-proliferative activity against CRC cells. This finding encourages further investigation into its anti-proliferative effects and the molecular mechanism involved. The results demonstrated that compound 225# effectively inhibited proliferation, arrested the cell cycle, induced cell apoptosis and promoted autophagy in CRC cells. First, the anticancer activity of compound 225# in CRC cells was investigated, along with its cytotoxicity on normal human colorectal epithelial cells. Notably, it was observed that compound 225# effectively inhibited the viability of CRC cells, while only slightly affecting that of normal human colorectal epithelial cells, indicating its high antitumor activity and low toxicity. In addition, the effects of compound 225# on the proliferation of CRC cells were assessed using colony formation and EdU assays. The results demonstrated a marked dose-dependent inhibition of colony formation and the number of EdU-positive cells in response to compound 225#. Second, the antitumor effects of compound 225# were evaluated in an in vivo xenograft mouse model, where it exhibited dose-dependent inhibition of tumor growth. Moreover, the weight loss observed in mice treated with compound 225# was not significantly higher than that in the control group, indicating its low toxicity. Based on the findings from both in vitro and in vivo studies, compound 225# shows promise as a therapeutic agent for CRC.
The dysregulation of cell cycle progression is a key characteristic of cancer. Inducing cell cycle arrest, especially in the G2/M phase, may be an effective strategy to address uncontrolled cancer cell proliferation (16). Cyclin B1 and cyclin A1, along with CDK1, serve as specific regulators in the G2/M phase, and they facilitate the division of one cell into two (17). Flow cytometric analysis revealed that compound 225# induced G2/M phase arrest in CRC cells. Further western blotting showed that treatment with compound 225# enhanced the expression levels of p-CHK2, p-ATR, p-BRCA1 and γ-H2A.X, while decreasing the expression levels of p-P53, indicating DNA damage to the cell cycle. It has previously been reported that downregulation of p-P53 is associated with DNA damage and CHK2 activation (18). P21 plays a critical role in blocking activation of CDK1/cyclin B1 in a P53-dependent or independent manner (19). Mechanistically, compound 225# treatment was revealed to cause a marked downregulation of SKP2. Two crucial tumor suppressors, P21 and P27 (20), which act as the substrate for SKP2, were found to be upregulated in compound 225#-treated CRC cells in the present study. The results of the present study revealed that compound 225# exerts a tumor suppressive effect, which is partly attributed to the inhibition of SKP2 and promotion of its downstream targets P21 and P27.
Apoptosis, which is one of the primary mechanisms of programmed cell death, serves a critical role in maintaining homeostasis in the human body (21). Induction of apoptosis in tumors is a key mechanism targeted by numerous anticancer drugs. Therefore, it is important to study the effects of anticancer agents that induce apoptosis. In the present study, the effects of compound 225# on apoptosis in CRC cells were detected by flow cytometry. The findings revealed that compound 225# markedly induced apoptosis in both HCT116 and SW620 CRC cells. Notably, PUMA, located downstream of the P53 gene, is a pro-apoptotic initiator gene, which can activate apoptosis through the P53-dependent pathway and thus induce apoptosis in tumor cells. PARP is broken down by caspases as a substrate to form cleaved PARP, which can ultimately lead to cell death. Compound 225# induced apoptosis in HCT116 and SW620 cells by increasing the expression levels of the proapoptotic protein PUMA and by cleaving PARP.
The ER is widely found in eukaryotic cells, and is responsible for protein synthesis, folding, assembly and transport, and drug metabolism. External environmental stimuli can lead to ER dysfunction in cells, and ER stress occurs via the accumulation of misfolded proteins, which is a self-protective mechanism (22). However a series of pathological responses can occur when the number of misfolded proteins exceeds the processing capacity of the ER (23). Generally, the level of intracellular molecular chaperones is correspondingly raised when ER stress occurs (24). Calnexin exists in the ER as a Ca2+-requiring lectin-like molecular chaperone protein, which binds to the oligosaccharide chains of newly synthesized proteins that have not been fully folded, preventing the proteins from aggregating and ubiquitinating with each other, and inhibiting incomplete proteins from leaving the ER (25). Notably, Ub protein aggregation is an abnormal manifestation of protein degradation. However, in the present study, the protein expression levels of calnexin were increased after treatment with compound 225#, indicating that the aggregation of ubiquitinated proteins and the spillover of incomplete folded proteins was not prevented, which was consistent with the results of the western blot detection of increased ubiquitinated protein expression.
Under ER stress conditions, to temporarily weaken mRNA translation at the overall level in the cell and to thus relieve the pressure of intracellular proteins, PERK forms dimers that are subsequently activated by self-phosphorylation, which further promotes the phosphorylation of downstream eIF2α (26). PDI serves not only as an enzyme that catalyzes the formation and isomerization of disulfide bonds, but also as an important chaperone (27). The amount of PDI increases with the increase of misfolded or unfolded proteins in the ER. Over the past several years, the potential of PDI as a drug target, particularly in cancer therapy, has received considerable attention (28). PDI plays a crucial role in facilitating the degradation of misfolded proteins via ER-associated degradation. This process involves the translocation of damaged proteins from the ER to the cytoplasm, where they are subsequently ubiquitinated and degraded by proteasome hydrolase (29). After treatment with compound 225#, in order to maintain cellular protein homeostasis and prevent protein misfolding and aggregation, the defenses of molecular chaperones are triggered and molecular chaperone expression is elevated, which in turn indicates the occurrence of ER stress response. Therefore, in the present study, compound 225# led to elevated protein expression levels of PDI, p-eIF2α and PERK. The imbalance of protein metabolism and ER stress in CRC cells shows promise to become one of the mechanisms that may inhibit the proliferation of CRC cells. Therefore, targeting the ER stress signaling pathway could be a promising strategy to treat CRC.
ER stress is a well-known cellular stress response that serves a crucial role in activating autophagy. Autophagy, a highly significant and evolutionarily conserved mechanism, is responsible for maintaining cellular homeostasis (15). Previous studies have demonstrated that the activation of autophagy is dependent on the induction of ER stress (30,31). The autophagy induced by ER stress can be evidenced by the increase of GFP-LC3 puncta and accumulation of LC3-II. LC3-II promotes the expansion and maturation of autophagy, which is considered a signal of autophagy activation (32).
GFP-mCherry-LC3B Tandem fluorescent protein is a fusion protein specifically used to detect the level of autophagic flux. When there is only red fluorescence and no green fluorescence in the cell, there is no overlap and therefore no yellow fluorescence, indicating that the fusion protein is located in the lysosome or autophagic lysosome, i.e. autophagic flux activation (33). This highlights its significance as a potential target for the development of anticancer drugs (34,35). Activating autophagy in cancer cells is being recognized as a promising strategy to improve the effectiveness of chemotherapy in cancer treatment. However, it is crucial to ensure that autophagy can be highly activated in cancer cells for it to be considered a viable therapeutic target (36). The present study demonstrated that induction of autophagy by compound 225# could inhibit the proliferative activity of CRC cells. The present research is a preliminary study on the anticancer effect of compound 225#. In conclusion, the present study provided evidence for the significant anticancer activity of compound 225# in CRC both in vitro and in vivo. Specifically, compound 225# inhibited the proliferation of CRC cells by inducing cell cycle arrest, apoptosis and autophagy. Notably, ER stress was revealed to contribute to the compound 225#-mediated pathological response in CRC cells. The findings of the present study suggested that compound 225# may have the potential to be an effective therapeutic agent or adjuvant for the treatment of CRC. While the present study provides evidence for the significant anticancer activity of compound 225# against CRC in vitro and in vivo, there are some limitations. Firstly, this study is only a preliminary study of the anticancer effects of compound 225#, and its exact mechanism of action is not fully understood and needs to be explored further. Secondly, other specific studies on this substance, such as combinations with existing anticancer drugs, have not yet been carried out, and this area could be further explored in future studies. Finally, although the present study revealed that ER stress may be one of the causes of the compound 225#-mediated pathological response in CRC cells, other possible mechanisms have not yet been fully elucidated, which again needs to be refined by further studies. Therefore, more in-depth and comprehensive studies on the anticancer effects of compound 225# and its mechanisms are still needed.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This research was supported by the Basic Research and Frontier Program of Chongqing Science and Technology Bureau (grant no. cstc2020jcyj-msxmX0733), the Science and Technology Research Program of Chongqing Municipal Education Commission (grant nos. KJQN202201329, KJQN202201330 and KJZD-K202001302), The Science and Technology Program of Yongchuan Science and Technology Bureau (grant nos. 2022yc-jckx20014 and 2021yc-jckx20044), the Science and Technology Research Program of Chongqing Education Commission (grant no. KJQN202201205), and the Graduate Scientific Research Innovation Project of Chongqing Three Gorges University (grant no. YJSKY21032).
Availability of data and materials
The data generated in the present study may be requested from the corresponding author.
Authors' contributions
XZ and LH performed the literature retrieval, designed and conducted the experiments, analyzed the data and drafted the manuscript. YL designed and synthesized compound 225#. YQ, WH and WL participated in experiments. HD and DY conceived and designed the experiments, rigorously revised the manuscript and finally approved the version to be submitted. LH and DY confirm the authenticity of all the raw data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All of the animal studies performed were reviewed and approved by the Ethics Committee for Animal Studies at Chongqing University of Arts and Sciences (approval no. CQWU202204; Chongqing, China). The experiments were strictly conducted according to standard protocols.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
American Cancer Society, . Cancer facts & figures 2023. Atlanta Ga: American Cancer Society; 2023 | |
|
Zang H, Yang W and Tian X: Simvastatin in the treatment of colorectal cancer: A review. Evid Based Complement Alternat Med. 2022:38279332022. View Article : Google Scholar : PubMed/NCBI | |
|
Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N, et al: Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 520:368–372. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Wu Q, Deng J, Fan D, Duan Z, Zhu C, Fu R and Wang S: Ginsenoside Rh4 induces apoptosis and autophagic cell death through activation of the ROS/JNK/p53 pathway in colorectal cancer cells. Biochem Pharmacol. 148:64–74. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Luo YR, Zhou ST, Yang L, Liu YP, Jiang SY, Dawuli Y, Hou YX, Zhou TX and Yang ZB: Porcine epidemic diarrhoea virus induces cell-cycle arrest through the DNA damage-signalling pathway. J Vet Res. 64:25–32. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Chien JH, Chang KF, Lee SC, Lee CJ, Chen YT, Lai HC, Lu YC and Tsai NM: Cedrol restricts the growth of colorectal cancer in vitro and in vivo by inducing cell cycle arrest and caspase-dependent apoptotic cell death. Int J Med Sci. 19:1953–1964. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Sun S, Yi Y, Xiao ZXJ and Chen H: ER stress activates TAp73α to promote colon cancer cell apoptosis via the PERK-ATF4 pathway. J Cancer. 14:1946–1955. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Geng J, Guo Y, Xie M, Li Z, Wang P, Zhu D, Li J and Cui X: Characteristics of endoplasmic reticulum stress in colorectal cancer for predicting prognosis and developing treatment options. Cancer Med. 12:12000–12017. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Lei Y, He L, Yan C, Wang Y and Lv G: PERK activation by CCT020312 chemosensitizes colorectal cancer through inducing apoptosis regulated by ER stress. Biochem Biophys Res Commun. 557:316–322. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Chen X and Cubillos-Ruiz JR: Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 21:71–88. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Wang M and Kaufman RJ: The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 14:581–597. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
He LJ, Yang DL, Li SQ, Zhang YJ, Tang Y, Lei J, Frett B, Lin HK, Li HY, Chen ZZ and Xu ZG: Facile construction of fused benzimidazole-isoquinolinones that induce cell-cycle arrest and apoptosis in colorectal cancer cells. Bioorgan Med Chem. 26:3899–3908. 2018. View Article : Google Scholar | |
|
Deka D, D'incà R, Sturniolo G C, Das A, Pathak S and Banerjee A: Role of ER stress mediated unfolded protein responses and ER stress inhibitors in the pathogenesis of inflammatory bowel disease. Digest Dis Sci. 67:5392–5406. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Xu Y and Fang D: Endoplasmic reticulum-associated degradation and beyond: The multitasking roles for HRD1 in immune regulation and autoimmunity. J Autoimmun. 109:1024232020. View Article : Google Scholar : PubMed/NCBI | |
|
Song S, Tan J, Miao Y and Zhang Q: Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J Cell Physiol. 233:3867–3874. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Wang G, Zhang T, Sun W, Wang H, Yin F, Wang Z, Zuo D, Sun M, Zhou Z, Lin B, et al: Arsenic sulfide induces apoptosis and autophagy through the activation of ROS/JNK and suppression of Akt/mTOR signaling pathways in osteosarcoma. Free Radical Bio Med. 106:24–37. 2017. View Article : Google Scholar | |
|
Xia Y, Lei Q, Zhu Y, Ye T, Wang N, Li G, Shi X, Liu Y, Shao B, Yin T, et al: SKLB316, a novel small-molecule inhibitor of cell-cycle progression, induces G2/M phase arrest and apoptosis in vitro and inhibits tumor growth in vivo. Cancer Lett. 355:297–309. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Ji YM, Zhang KH, Pan ZN, Ju JQ, Zhang HL, Liu JC, Wang Y and Sun SC: High-dose zearalenone exposure disturbs G2/M transition during mouse oocyte maturation. Reprod Toxicol. 110:172–179. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang C, Liu K, Yao K, Reddy K, Zhang Y, Fu Y, Yang G, Zykova TA, Shin SH, Li H, et al: HOI-02 induces apoptosis and G2-M arrest in esophageal cancer mediated by ROS. Cell Death Dis. 6:e19122015. View Article : Google Scholar : PubMed/NCBI | |
|
Ye D, Li Z and Wei C: Genistein inhibits the S-phase kinase-associated protein 2 expression in breast cancer cells. Exp Ther Med. 15:1069–1075. 2018.PubMed/NCBI | |
|
Gary AS and Rochette PJ: Apoptosis, the only cell death pathway that can be measured in human diploid dermal fibroblasts following lethal UVB irradiation. Sci Rep. 10:189462020. View Article : Google Scholar : PubMed/NCBI | |
|
Sims SG, Cisney RN, Lipscomb MM and Meares GP: The role of endoplasmic reticulum stress in astrocytes. Glia. 70:5–19. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Carreras-Sureda A, Pihán P and Hetz C: The unfolded protein response: At the intersection between endoplasmic reticulum function and mitochondrial bioenergetics. Front Oncol. 7:552017. View Article : Google Scholar : PubMed/NCBI | |
|
Gardner BM, Pincus D, Gotthardt K, Gallagher CM and Walter P: Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol. 5:a0131692013. View Article : Google Scholar : PubMed/NCBI | |
|
Bergeron JJ, Brenner MB, Thomas DY and Williams DB: Calnexin: A membrane-bound chaperone of the endoplasmic reticulum. Trends Biochem Sci. 19:124–128. 1994. View Article : Google Scholar : PubMed/NCBI | |
|
Sundaram A, Appathurai S, Plumb R and Mariappan M: Dynamic changes in complexes of IRE1α, PERK, and ATF6α during endoplasmic reticulum stress. Mol Biol Cell. 29:1376–1388. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Kamarehei M, Pejman S, Ardestani SK, Zahednasab H, Firouzi M and Harirchian MH: Inhibition of protein disulfide isomerase has neuroprotective effects in a mouse model of experimental autoimmune encephalomyelitis. Int Immunopharmacol. 82:1062862020. View Article : Google Scholar : PubMed/NCBI | |
|
Kurpińska A, Suraj-Prażmowska J, Stojak M, Jarosz J, Mateuszuk Ł, Niedzielska-Andres E, Smolik M, Wietrzyk J, Kalvins I, Walczak M and Chłopicki S: Comparison of anti-cancer effects of novel protein disulphide isomerase (PDI) inhibitors in breast cancer cells characterized by high and low PDIA17 expression. Cancer Cell Int. 22:2182022. View Article : Google Scholar : PubMed/NCBI | |
|
Dunlop R, Powell J, Metcalf J, Guillemin G and Cox P: L-serine-mediated neuroprotection includes the upregulation of the er stress chaperone protein disulfide isomerase (PDI). Neurotox Res. 33:113–122. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Chandrika BB, Yang C, Ou Y, Feng X, Muhoza D, Holmes AF, Theus S, Deshmukh S, Haun RS and Kaushal GP: Endoplasmic reticulum stress-induced autophagy provides cytoprotection from chemical hypoxia and oxidant injury and ameliorates renal ischemia-reperfusion injury. PLoS One. 10:e01400252015. View Article : Google Scholar : PubMed/NCBI | |
|
Bao Y, Pu Y, Yu X, Gregory BD, Srivastava R, Howell SH and Bassham DC: IRE1B degrades RNAs encoding proteins that interfere with the induction of autophagy by ER stress in Arabidopsis thaliana. Autophagy. 14:1562–1573. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Li JJ, Yan YY, Sun HM, Liu Y, Su CY, Chen HB and Zhang JY: Anti-cancer effects of pristimerin and the mechanisms: A critical review. Front Pharmacol. 10:7462019. View Article : Google Scholar : PubMed/NCBI | |
|
Nyfeler B, Bergman P, Triantafellow E, Wilson CJ, Zhu Y, Radetich B, Finan PM, Klionsky DJ and Murphy LO: Relieving autophagy and 4EBP1 from rapamycin resistance. Mol Cell Biol. 31:2867–2876. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Yang C, Yang QO, Kong QJ, Yuan W and Yang YPO: Parthenolide induces reactive oxygen species-mediated autophagic cell death in human osteosarcoma cells. Cell Physiol Biochem. 40:146–154. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Ravanan P, Srikumar IF and Talwar P: Autophagy: The spotlight for cellular stress responses. Life Sci. 188:53–67. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Xie CM, Chan WY, Yu S, Zhao J and Cheng CHK: Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radical Bio Med. 51:1365–1375. 2011. View Article : Google Scholar : PubMed/NCBI |